
Chapter 9 – Reaction Energetics
Introduction
We use the term energetics to combine two very important fields of study: thermodynamics and kinetics. Thermodynamics is the study of energy and its transformations. Kinetics is the study of the rates and mechanisms of reactions. A thermodynamic study of a reaction examines energy differences between the reactants and products. These studies allow us to predict how much product can be obtained. Thermodynamics is not concerned with how long the reaction takes or how it occurs. These latter concerns are the domain of kinetics. In this chapter, we examine the energetics of some reactions to better understand how reactions occur, to learn how to predict the feasibility of a reaction, and to determine the effect of temperature on both the amount of product and the rate of reaction.9.1 First Law of Thermodynamics
Introduction
Thermodynamics is the study of energy and its transformations. In chemistry, thermodynamics studies consider the energy difference between the reactants and products. These studies allow us to predict the amount of product that we can expect. We start our study of thermodynamics with the first law of thermodynamics—energy cannot be created or destroyed.Objectives
-
•Predict the sign of ΔE for common processes.
-
•Indicate the direction of energy flow from the sign of ΔE.
-
•Distinguish between system, surroundings, and universe.
-
•State the first law of thermodynamics.
-
•Define the terms q and w and explain what their signs indicate.
-
•Distinguish between endothermic and exothermic.
-
•Determine the energy change of a system given the amount of heat that it absorbs or gives off and the amount of work it does or has done on it.
9.1-1. Energy Change
The sign of ΔE indicates the direction of energy flow.
ΔE = Efinal − Einitial.
-
•ΔE > 0means that the energy of the final state of the system is greater than that of the initial state, so this energy flows into the system.
-
•ΔE < 0means that the energy of the final state of the system is less than that of the initial state, so this energy flow out of the system.
-
•ΔE = 0means that the energy of the final and initial states of the system are the same, so there is no energy flow.
9.1-2. Sign of ΔE Exercise
Exercise 9.1:
Indicate whether
ΔE < 0,
ΔE > 0,
or ΔE = 0
for each of the following. Assume that the system is the object mentioned in the problem.
-
heating water 25 °C to 50 °C
-
ΔE < 0 The energy of an object always increases with temperature, so ΔE > 0.
-
ΔE > 0
-
ΔE = 0 The energy of an object always increases with temperature, so ΔE > 0.
-
moving positive charge closer to negative charge
-
ΔE < 0
-
ΔE > 0 Opposite charges attract because their energy is reduced in doing so. Thus, ΔE < 0.
-
ΔE = 0 Opposite charges attract because their energy is reduced in doing so. Thus, ΔE < 0.
-
evaporating 1 g of water at 25 °C
-
ΔE < 0 Recall from Chapter 8 that the energy of the gaseous state is much greater that of than the liquid state, so evaporation always involves an increase in energy, so ΔEvap > 0.
-
ΔE > 0
-
ΔE = 0 Recall from Chapter 8 that the energy of the gaseous state is much greater that of than the liquid state, so evaporation always involves an increase in energy, so ΔEvap > 0.
-
moving a ball from a table top to the floor
-
ΔE < 0
-
ΔE > 0 The potential energy of the ball is decreased due to the gravitational field of the earth, so ΔE < 0.
-
ΔE = 0 The potential energy of the ball is decreased due to the gravitational field of the earth, so ΔE < 0.
-
heating a piece of zinc from 25 °C to 75 °C and then cooling it back to
-
ΔE < 0 The initial and final temperatures are the same, so Efinal = Einitial. Therefore, ΔE = Efinal – Einitial = 0.
-
ΔE > 0 The initial and final temperatures are the same, so Efinal = Einitial. Therefore, ΔE = Efinal – Einitial = 0.
-
ΔE = 0
25 °C
9.1-3. First Law
Energy can be moved from one place to another, but it cannot be created or destroyed.
ΔEuniv = ΔE + ΔEsur
ΔEuniv = 0
implies the following.
We conclude that the first law of thermodynamics implies that the energy of a system can be changed only by exchanging energy with its surroundings.
9.1-4. Determining ΔE Exericse
Exercise 9.2:
75 J of energy is transferred when a hot rod is cooled in water. Determine the value of ΔE for the process when each of the following are considered to be the system.
rod
-75_0__
The rod cools, so it gives up energy, i.e., its energy decreases. ΔE = –75 J if the rod is the system.
J
water
75_0__
The 75 J of energy that is lost by the rod must enter the water, so the energy of the water increases. Thus, ΔE = +75 J if the water is the system.
J
water and rod
0_0__
If energy is only transferred between the water and the rod and none enters or leaves the system from its surroundings, then ΔE = 0.
J
A reaction releases 175 kJ of energy. Answer the following if the reaction is the system.
ΔE =
-175_0__
The system releases heat, so Efinal < Einitial, which means that ΔE < 0. Consequently, ΔE = –175 kJ.
kJ
ΔEsur =
175_0__
ΔEsur = –ΔE = –(–175 kJ) = +175 kJ. The surroundings must absorb the heat released by the system.
kJ
ΔEuniv =
0_0__
ΔEuniv = 0 for all processes.
kJ
9.1-5. Heat and Work
When q and w are positive, energy flows into the system.

Table 9.1: The Signs of Heat and Work and the Direction of Energy Flow
9.1-6. Heat and Work Exercise
Exercise 9.3:
A system absorbs 500 J of heat and does 300 J of work.
What is the value of q?
q =
500_0__
System absorbs 500 J of heat, which increases its energy increases by 500 J.
q = +500 J – an endothermic process
J
What is the value of w?
w =
-300_0__
The system does 300 J of work, so its energy drops by 300 J.
w = –300 J
J
What is the energy change for the process?
ΔE =
200_0__
ΔE = q + w = +500 – 300 = +200 J
The energy of the system increases by 200 J. J
The energy of the system increases by 200 J. J
9.2 Enthalpy
Introduction
Typically, reactions are carried out at constant pressure and the initial and final temperatures are the same. Gases that are produced by a reaction that is open to the atmosphere can carry away some of the energy of the reaction (they do work when they escape), and that energy is lost. Thus, the thermodynamic property of a reaction that is carried out at constant pressure that is most important is the heat that is exchanged with the surroundings. In this section, we define that thermodynamic property.Objectives
-
•Define the enthalpy (heat) of reaction.
-
•Determine the relative energies of the reactants and products given the enthalpy of reaction.
-
•Define the standard state.
-
•Explain the meaning of a superscript zero on a thermodynamic quantity.
-
•Explain what the s, l, or g after each substance in a thermochemical equation specifies.
-
•Determine the enthalpy change for the reaction of a given number of moles of a reactant given the thermochemical equation for the reaction.
-
•Determine the enthalpy change of a reaction given the enthalpy change for the reaction in which the coefficients have been multiplied by some number and/or the reaction is written in the reverse direction.
9.2-1. Enthalpy of Reaction
The enthalpy of reaction is the heat absorbed at constant T and P. It is positive for endothermic reactions and negative for exothermic reactions.
The reactants are at higher energy than the products in exothermic reactions.

Figure 9.1a: Exothermic Processes
ΔH < 0
for the reaction. The reaction is exothermic because it gives off heat to the surroundings causing the surroundings to warm.
The reactants are at lower energy than the products in endothermic reactions.

Figure 9.1b: Endothermic Processes
ΔH > 0
for the reaction. The reaction is endothermic because it absorbs heat from the surroundings causing the surroundings to cool.
9.2-2. Standard States
Standard states are defined at 1 atm pressure, but there is no standard temperature.
-
•The standard state of a pure substance at a given temperature is its most stable form at that temperature and 1 atm pressure. Unless indicated differently, assume that solids and liquids are pure.
-
•Gases are considered pure substances even when there is a mixture of gases because the molecules are far apart and do not interact. Thus, the standard state of a gas is a partial pressure of 1 atm.
-
•The standard state of a dissolved substance is a concentration of 1 mole of substance per liter of solution at a pressure of 1 atm.
Exercise 9.4:
Indicate the standard state of each of the following.
-
oxygen at –20 °C
-
solid O2 is a gas at –20 °C. The standard state of gases is 1 atm pressure, so the standard state of O2 at –20 °C is 1 atm of gas.
-
liquid O2 is a gas at –20 °C. The standard state of gases is 1 atm pressure, so the standard state of O2 at –20 °C is 1 atm of gas.
-
1 atm gas
-
1 M concentration O2 is a gas at –20 °C. The standard state of gases is 1 atm pressure, so the standard state of O2 at –20 °C is 1 atm of gas.
-
water at –20 °C
-
solid
-
liquid The stable form of H2O at –20 °C is the solid, so ice is the standard state of H2O at –20 °C.
-
1 atm gas The stable form of H2O at –20 °C is the solid, so ice is the standard state of H2O at –20 °C.
-
1 M concentration The stable form of H2O at –20 °C is the solid, so ice is the standard state of H2O at –20 °C.
-
water at +20 °C
-
solid The stable form of H2O at +20 °C is the liquid, so liquid water is the standard state of H2O at +20 °C.
-
liquid
-
1 atm gas The stable form of H2O at +20 °C is the liquid, so liquid water is the standard state of H2O at +20 °C.
-
1 M concentration The stable form of H2O at +20 °C is the liquid, so liquid water is the standard state of H2O at +20 °C.
-
table sugar (C12H22O11) at 20 °C
-
solid
-
liquid The stable form of C12H22O11 at 20 °C is the solid, so C12H22O11 crystals are the standard state of C12H22O11 at 20 °C.
-
1 atm gas The stable form of C12H22O11 at 20 °C is the solid, so C12H22O11 crystals are the standard state of C12H22O11 at 20 °C.
-
1 M concentration The stable form of C12H22O11 at 20 °C is the solid, so C12H22O11 crystals are the standard state of C12H22O11 at 20 °C.
-
table sugar dissolved in water at
-
solid The standard state of a dissolved substance is a solution in which its concentration is 1 mol per liter. Therefore, the standard state of a solution of C12H22O11 is a solution in which there are 342 g (1 mole) of C12H22O11 per liter of solution.
-
liquid The standard state of a dissolved substance is a solution in which its concentration is 1 mol per liter. Therefore, the standard state of a solution of C12H22O11 is a solution in which there are 342 g (1 mole) of C12H22O11 per liter of solution.
-
1 atm gas The standard state of a dissolved substance is a solution in which its concentration is 1 mol per liter. Therefore, the standard state of a solution of C12H22O11 is a solution in which there are 342 g (1 mole) of C12H22O11 per liter of solution.
-
1 M concentration
20 °C
9.2-3. Standard Enthalpy of Reaction
The enthalpy change for a reaction in which all reactants and products are in their standard states is called the standard enthalpy of reaction and given the symbol ΔH° (the superscript '°' implies standard state conditions). Most enthalpies of reaction used in this course are standard enthalpies. A thermochemical equation is a balanced chemical equation that includes a thermodynamic property such asΔH°. Enthalpy changes depend upon the states of the substances, so the state is indicated by the following in parentheses.
-
•s = solid
-
•l = liquid
-
•g = gas
-
•aq = aqueous (water) solution
2 Na(s) + 2 H2O(l) → 2 NaOH(aq) + H2(g) ΔH° = –368 kJ
-
•two moles of solid sodium react,
-
•two moles of liquid water react,
-
•two moles of NaOH are produced in an aqueous solution, and
-
•one mole of H2 gas is produced.
9.2-4. Stoichiometry and Thermodynamics
The thermochemical equation,P4(s) + 6 Cl2(g) → 4 PCl3(g) ΔH° = –1152 kJ
| −1152 kJ |
| 1 mol P4 |
| −1152 kJ |
| 6 mol Cl2 |
| −1152 kJ |
| 4 mol PCl3 |
4 mol Cl2 ×
= −768 kJ
| −1152 kJ |
| 6 mol Cl2 |
9.2-5. Stoichiometry Exercise
Exercise 9.5:
Consider the thermochemical equation.
What would be the value of ΔH° under the following circumstances.
N2(g) + 3 H2(g) → 2 NH3(g) ΔH° = –92 kJ
one mole of ammonia is formed
-46___
The thermochemical equation indicates that 92 kJ of heat are released (minus sign) when 2 moles of NH3 are formed. Thus,
46 kJ would be released.
kJ
1 mol NH3 ×
= −46 kJ.
| −92 kJ |
| 2 mol NH3 |
two moles of H2 react
-61.3333___
The thermochemical equation indicates that 92 kJ of heat are released (minus sign) when 3 moles of H2 react. Thus,
61 kJ would be released.
kJ
2 mol H2 ×
= −61 kJ.
| −92 kJ |
| 3 mol H2 |
7.0 g of N2 react
-23___
The thermochemical equation indicates that 92 kJ of heat are released (minus sign) when 1 mole of N2 reacts. Thus, ΔH° = (–92 kJ)/(1 mol N2), which can be multiplied by the number of moles of N2 to obtain the answer.
23 kJ would be released.
kJ
7.0 g N2 ×
= 0.25 mol N2
| 1 mol N2 |
| 28 g N2 |
0.25 mol N2 ×
= −23 kJ
| −92 kJ |
| 1 mol N2 |
9.2-6. Properties of Enthalpy
We have seen that ΔH is proportional to the number of moles of substance that react. This means the following.
If the coefficients of a chemical equation are multiplied by some number, then the enthalpy of the resulting reaction equals the enthalpy of the original reaction multiplied by the same number.
H2O(s) → H2O(l) ΔH° = +6 kJ
2 H2O(s) → 2 H2O(l) ΔH° = +12 kJ
2 H2O(s) → 2 H2O(l) ΔH° = +12 kJ
The sign of ΔH is changed when the direction of the reaction is reversed.
H2O(s) → H2O(l) ΔH° = +6 kJ
H2O(l) → H2O(s) ΔH° = –6 kJ
H2O(l) → H2O(s) ΔH° = –6 kJ
1 mole of liquid water releases 6 kJ of heat.
Exercise 9.6:
The following thermochemical equation for burning butane is given.
Determine the enthalpy of reaction for each of the following.
C4H10(l) + 6.5 O2(g) → 4 CO2(g) + 5 H2O(l) Δ H° = –2855 kJ
2 C4H10(l) + 13 O2(g) → 8 CO2(g) + 10 H2O(l)
-5710___
The reaction involves twice as much butane, so the enthalpy change of the given reaction must be multiplied by two:
ΔH = 2(−2855) = −5710 kJ.
The resulting thermochemical equation is 2 C4H10(l) + 13 O2(g) → 8 CO2(g) + 10 H2O(l) ΔH° = –5710 kJ.
kJ
4 CO2(g) + 5 H2O(l) → C4H10(l) + 6.5 O2(g)
2855___
The reaction is simply the reverse of the original reaction, so the sign of the enthalpy change of the given reaction must be changed:
ΔH = −(−2855) = +2855 kJ.
The resulting thermochemical equation is 4 CO2(g) + 5 H2O(l) → C4H10(l) + 6.5 O2(g) ΔH° = +2855 kJ.
kJ
8 CO2(g) + 10 H2O(l) → 2 C4H10(l) + 13 O2(g)
5710___
The reaction involves twice the amounts in the reverse of the original reaction, so the given enthalpy change must be multiplied by two and the sign must be changed, i.e., the original value must be multiplied by –2:
ΔH = (−2)(−2855) = +5710 kJ.
The resulting thermochemical equation is 8 CO2(g) + 10 H2O(l) → 2 C4H10(l) + 13 O2(g) ΔH° = +5710 kJ.
kJ
9.3 Enthalpies of Combustion
Introduction
Most of the energy utilized by humans comes from combustion reactions (reactions with O2). The internal combustion engine that runs our cars gets its energy from the combustion of gasoline. Many homes are heated by the combustion of coal, oil, or natural gas. Many power plants generate electricity by burning (combusting) coal, and humans obtain the energy needed to sustain life by the combustion of carbohydrates. The carbon in the fuel is converted to CO2 and the hydrogen is converted to H2O by the reaction with O2. In this section, we examine enthalpies (heats) of combustion.Objectives
-
•Define enthalpy of combustion and write the chemical equation to which the enthalpy of combustion of a substance applies.
9.3-1. Enthalpies of Combustion
Combustion is the reaction of a substance with oxygen, and it is the major source of harnessed energy. Combustion reactions are always exothermic, and it is the energy released in combustion reactions that fuels our vehicles, heats our homes, and powers our bodies. The products of combustion are usually, but not always, the oxides of the elements comprising the molecule being combusted.
The standard molar enthalpy of combustion of a substance is the heat absorbed when one mole of the substance reacts with O2 at standard conditions.

Table 9.2: Thermochemical Equations for Some Common Combustion Reactions
9.3-2. Combustion Exercise
Exercise 9.7:
The calorie content of carbohydrates, fats, and proteins are all derived from their heats of combustion because they are all burned in the body. In this example, we use the heat of combustion of sugar to determine the number of calories in a teaspoon of sugar. The thermochemical equation for the combustion of sucrose (table sugar) is the following.
How many calories are in a heaping teaspoon of sugar (4.8 g)?
C12H22O11(s) + 12 O2(g) → 12 CO2(g) + 11 H2O(l) ΔH° = –5650 kJ/mol
What is the number of moles of sugar present (Mm = 342 g/mol)?
0.014___
4.8 g C12H22O11 ×
= 0.014 mol C12H22O11
mol
| 1 mol C12H22O11 |
| 342 g C12H22O11 |
What is the amount of heat liberated during the combustion?
79.298___
0.014 mol ×
= 79 kJ released
kJ
| 5650 kJ released |
| 1 mol |
If there are 4.18 kJ in a dietary calorie, what is the number of dietary calories present in the sugar?
18.97___
79 kJ ×
= 19 Cal
Cal
| 1 Cal |
| 4.18 kJ |
9.4 Bond Energies
Introduction
Recall from Chapter 5 that bond formation involves a lowering of the energy of the system, i.e., bond formation is an exothermic process. Reversing the direction of the reaction, changes the sign of ΔH, so bond breaking is an endothermic process. The energy released or absorbed during a chemical reaction comes from or is stored in chemical bonds, and the enthalpy change of a reaction can be estimated from tabulated values of bond energies.Prerequisites
Objectives
-
•Define the bond or dissociation energy.
-
•Estimate the enthalpy of reaction from bond energies.
9.4-1. Bond Energies
The bond energy or dissociation energy (D) of a bond is the energy required to break one mole of bonds in the gas phase.

Table 9.3: Examples of Some Thermochemical Equations involving Bond Energies
9.4-2. Estimating Enthalpy
Reactions involve breaking and forming bonds, and the heat generated or absorbed during a reaction is the net result. If we knew the energies of all of the interactions involved, we could determine the enthalpy of reaction by determining how much energy must be supplied to break all of the interactions that had to be broken and subtracting the energy that is released when all of the new interactions form. Bond energies give us estimates of these energies, but tabulated bond energies are averages. For example, a C-Cl bond in CCl4 is different from a C-Cl bond in COCl2, but only one C-Cl bond energy is tabulated. In addition, bond energies are defined only for gas phase molecules, so other interactions can be ignored, but many reactions occur in solution where solvation and other interactions complicate the problem. However, bond energies can be used to obtain approximate enthalpies from the following.
The enthalpy of a reaction can be approximated by the sum of the bond energies of the bonds that must be broken less the sum of the bond energies of those that must be formed.
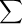
Example:
For example, let us approximate the enthalpy of reaction for the following.
One H-H bond and one F-F bond must be broken and two H-F bonds must be formed. The needed bond energies are given in Table 9.3.
For example, let us approximate the enthalpy of reaction for the following.
H2(g) + F2(g) → 2 HF(g)
| ΔH | ~ |
| ||||||||
| ~ | DH-H + DF-F − 2DH-F | |||||||||
| = | 436 + 155 − 2(565) | |||||||||
| = | −539 kJ |

9.4-3. Bond Energy Exercise
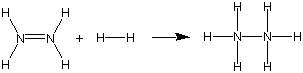
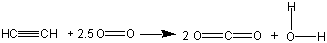
Exercise 9.8:
9.4-4. Bond Energy and Lewis Structure Exercise
Exercise 9.9:
Determine ΔH for the following reaction.
First determine the Lewis structures to get the bond orders of all bonds. Review Section 5.6 Determining Lewis Structures if you have forgotten how to determine Lewis structures.
H2CO(g) + NH3(g) → H2CNH(g) + H2O(g)
Bond Orders: All bonds to hydrogen are single bonds, but the other bond orders need to be determined.
C-O bond order in H2CO
2___
There are 4 shared pairs in H2CO, 2 are used for C-H bonds, so 2 are in the C-O bond.
C-N bond order in H2CNH
2___
There are 5 shared pairs in H2CNH. Three are used for CH and NH bonds, so 2 are in the C-N bond.
Enthalpies: Use the Bond Energies resource to determine the following enthalpies. Break and form only those bonds that do not appear in both the reactant and product side.
energy required to break the reactant bonds
1581___
The two C-H bonds and one N-H bond appear on both sides of the equation, so they are not included in the calculation.
D(C=O) + 2D(N-H) = 799 + 2(391) = 1581 kJ kJ
D(C=O) + 2D(N-H) = 799 + 2(391) = 1581 kJ kJ
energy released when product bonds are formed
1541___
The two C-H bonds and one N-H bond appear on both sides of the equation, so they are not included in the calculation.
D(C=N) + 2D(O-H) = 615 + 2(463) = 1541 kJ kJ
D(C=N) + 2D(O-H) = 615 + 2(463) = 1541 kJ kJ
Finally, determine the enthalpy of reaction.
ΔH =
40___
Energy required to break reactant bonds – energy released when product bonds are formed = 1581 – 1541 = 40 kJ.
kJ
9.5 Entropy
Introduction
A spontaneous process is one that takes place without intervention. A ball rolls down a hill spontaneously, but it does not roll uphill spontaneously. It can be made to roll uphill but only with intervention. We have used the concept that systems in nature strive to lower energy, so our first hypothesis might be that exothermic processes are spontaneous, while endothermic processes are not. However, there are spontaneous endothermic processes, such as evaporation. We saw in Chapter 8 that the energy of the molecules in the gas phase is much greater than those in the liquid phase, yet liquids do evaporate spontaneously, i.e., molecules go spontaneously from the lower energy liquid state into the higher energy gas phase. Clearly, there is another factor, and, in this section, we consider that factor.Objectives
-
•Distinguish between translational, rotational, and vibrational motion.
-
•Explain how the entropy of a system depends upon how freely its molecules move.
-
•Define entropy.
-
•Determine whether the entropy of reaction is positive, negative, or very small.
-
•State the relationship between the entropy of a system and the order of a system.
-
•Indicate the effects on the entropy of a system caused by adding heat and how the effect is dependent upon the temperature at which the heat is added .
9.5-1. Probability and Constraints
Unconstrained systems are statistically favored over constrained ones.
10! = 3,628,800
ways to arrange ten books on a shelf, but the books are aphabetized in only one. Thus, there are 3,628,799 ways to disburse the books in a random or disordered manner, but only one in which the books are constrained to be in alphabetical order. If the books were alphabetized originally but then removed and returned to the shelf without the intervention of a librarian, they would spontaneously move to a disordered state for strictly statistical reasons. If we assume that there are only two systems in our set of books, constrained or ordered and unconstrained or disordered, then the process constrained → unconstrained is spontaneous because there are more ways in which the books can be disbursed in the unconstrained, disordered system. The process unconstrained → constrained is not spontaneous and occurs only with intervention (a librarian in this example).
9.5-2. Types of Motion
The kinetic energy of a molecule is a combination of translational, rotational, and vibrational contributions.
-
•translation: straight line motion of the entire molecule through space
-
•rotation: circular motion of the atoms in a molecule about an axis fixed to the molecule
-
•vibration: atoms within a molecule moving relative to one another by causing small changes in bond lengths and bond angles

Figure 9.2: Degrees of Freedom of a Diatomic Molecule
-
aMotion of both atoms in the same direction translates the molecule in that direction.
-
bMotion in opposite directions along the x- or y-axis rotates the molecule about the y- or x-axis, respectively.
-
cMovement in opposite directions along a bonding axis results in a vibration as the bond elongates and contracts. A vibration is referred to with the greek symbol nu (ν).
9.5-3. Entropy
Entropy is a measure of the number of ways that a system can distribute its energy.
-
•Systems with higher entropies can distribute energy in more different ways than those with lower entropies.
Sgas >> Sliquid > Ssolid
9.5-4. Systems Strive to Increase Their Entropy
The two driving forces behind a process are the tendencies for systems to minimize their potential energy(ΔH < 0)
and to maximize the number of ways in which they can distribute their energy (ΔS > 0).
However, unless the entropy change is large, the tendency to minimize enthalpy is the dominant driving force. We have seen how to estimate enthalpy changes from bond energies, and we now examine how to predict whether the entropy change is large and positive, large and negative, or negligible.
The entropy change of a reaction is given as ΔS = Sproducts − Sreactants,
so ΔS > 0
when the entropy of the products is greater than that of the reactants. At this point, we need only predict whether ΔS is large, and, if so, what is its sign. To do this, we recall that Sgas >> Sliquid > Ssolid.
Indeed, we will assume that Sliquid ~ Ssolid ~ 0 to conclude the following.
Entropy changes are large only in reactions in which the number of moles of gas changes.
-
•ΔS > 0 for reactions that produce gas.
-
•ΔS < 0 for reactions that consume gas
-
•ΔS ~ 0 (is usually negligibly small) for reactions that do not involve a change in the number of moles of gas.
9.5-5. Constraints and Disorder as Predictors of Entropy
Constrained systems are more ordered and have lower entropies than systems that are less constrained.
-
•The more disordered a system is, the greater is its entropy.
9.5-6. Entropy Exercise
Exercise 9.10:
Predict which system in each pair has the greater entropy.
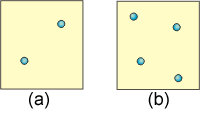
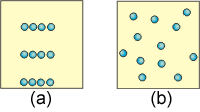
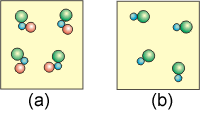
9.5-7. Effects of Heat and Temperature on Entropy
The number of ways a system can distribute its energy increases with the amount of energy it has to distribute, so the entropy of a system always increases with temperature. Similarly, adding heat to a system always increases the entropy of a system. However, 1 J of heat has much more impact when it is added to a system that has less energy (lower T) than it does when added to a system that has more energy (higher T). This relationship is expressed in the following equation, which defines entropy change. qrev is used to indicate that the heat must be added reversibly, which means that it must be added so slowly that the slightest change could reverse the direction of heat flow. Equation 9.5 can be rearranged toTΔS = qrev,
so TΔS is an energy term.
Using order as a predictor of entropy, we can understand the above with a simple analogy: the effect on the order of a room resulting when a book is casually dropped in it. The probability is very high that the book will NOT drop into its allotted location, so the dropped book will increase the disorder (entropy) of the system. In a neat room (representing a more ordered, lower temperature system), the effect of the out-of-place book is dramatic. However, its effect on a messy room (representing a higher temperature, less ordered system) is negligible as the out-of-place book is hardly noticeable. Similarly, adding 1 J of heat to a solid at 5 K (very ordered, low entropy) has a much more dramatic effect on the entropy than adding 1 J to the vapor at 500 K (disordered, high entropy).
9.5-8. Predicting Entropy Change Example
Exercise 9.11:
Predict the sign of ΔS° for each of the following reactions.
-
CaCO3(s) → CaO(s) + CO2(g)
-
ΔS < 0The reaction produces gas, so ΔS° > 0. The actual value is 159 J/K.
-
ΔS > 0
-
ΔS ~ 0The reaction produces gas, so ΔS° > 0. The actual value is 159 J/K.
-
3 H2(g) + N2(g) → 2 NH3(g)
-
ΔS < 0
-
ΔS > 0The balanced equation shows four moles of gas on the right and only two on the left. Consequently, gas is consumed, so ΔS° < 0. The actual value is –199 J/K.
-
ΔS ~ 0The balanced equation shows four moles of gas on the right and only two on the left. Consequently, gas is consumed, so ΔS° < 0. The actual value is –199 J/K.
-
Ag(s) + NaCl(s) → AgCl(s) + Na(s)
-
ΔS < 0There are no gases involved in the reaction, so the entropy change is expected to be small, i.e., ΔS° ~ 0. The actual value is 33 J/K.
-
ΔS > 0There are no gases involved in the reaction, so the entropy change is expected to be small, i.e., ΔS° ~ 0. The actual value is 33 J/K.
-
ΔS ~ 0
-
H2O(l) → H2O(g)
-
ΔS < 0The reaction produces gas, so ΔS
-
ΔS > 0
-
ΔS ~ 0The reaction produces gas, so ΔS
-
H2O(l) at 25 °C → H2O(l) at 5 °C
-
ΔS < 0
-
ΔS > 0The entropy of a system always decreases when its temperature is lowered. Consequently, ΔS° < 0, but it is small because the temperature change is small. The actual value is –13 J/K.
-
ΔS ~ 0The entropy of a system always decreases when its temperature is lowered. Consequently, ΔS° < 0, but it is small because the temperature change is small. The actual value is –13 J/K.
9.5-9. Entropy Change Example
Exercise 9.12:
Indicate which process in each of the following pairs increases the entropy of the system more.
-
adding 10 J of heat to neon at 300 K Use ΔS = q/T. The heat is added at the same temperature, so the greater entropy change occurs when the greater amount of heat is added. Consequently, adding 50 J results in a greater entropy change than adding 10 J at 300 K.
-
adding 50 J of heat to neon at 300 K
-
adding 10 J of heat to neon at 300 K
-
adding 10 J of heat to neon at 800 K Use ΔS = q/T. The amount of heat added is the same, so the greater entropy change occurs when the heat is added at the lower temperature. Consequently, adding 10 J results in a greater entropy change at 300 K.
-
adding 50 J of heat to neon at 800 K Use ΔS = q/T. (50/800) > (10/300), so adding 50 J at 800 K causes a larger entropy change.
-
adding 10 J of heat to neon at 300 K Use ΔS = q/T. (50/800) > (10/300), so adding 50 J at 800 K causes a larger entropy change.
-
melting 1 g of neon at its melting point Use ΔS = Sfinal – Sinitial.
- melting: ΔS = Sliquid – Ssolid
- evaporating: ΔS = Sgas – Sliquid
-
evaporating 1 g of neon at its boiling point
9.6 Second Law of Thermodynamics
Introduction
The fact that systems seek to increase their entropy is the basis of the Second Law of Thermodynamics, the topic of this chapter.Objectives
-
•State the Second Law of Thermodynamics.
9.6-1. The Second Law of Thermodynamics
Spontaneous processes must increase the entropy of the universe.
-
•Second Law of Thermodynamics: The entropy of the universe increases in all spontaneous processes; that is ΔSuniv > 0 for all spontaneous processes.
9.6-2. Entropy and Heat Flow
Heat flows spontaneously from hot to cold, never cold to hot. (It is forced to flow cold to hot in a heat pump, but energy is required, so it is not spontaneous.) We now show that this is a consequence of the second law. Consider to thermal resevoirs at T1 and T2 that are in thermal contact as shown below.
Figure 9.3
ΔSuniv = ΔS1 + ΔS2 > 0
ΔS =
=
J/K
.
| heat added reversibly |
| temperature at which the heat is added |
| qrev |
| T |
ΔS1 =
ΔS2 =
| −q |
| T1 |
| +q |
| T2 |
ΔSuniv =
+
| −q |
| T1 |
| +q |
| T2 |
ΔSuniv > 0
and the process is spontaneous only if T1 > T2.
In other words, only if the heat flows from the hotter sources to a cooler one.
9.7 Free Energy
Introduction
We now show that ΔSuniv can be written as a sum of system quantities, and, in doing so, derive a new thermodynamic property that includes the effects of both ΔH and ΔS on the spontaneity of any process carried out at constant temperature and pressure. The new property is called the free energy because it is the energy that is free to do work.Objectives
-
•Define Gibbs free energy, and explain why it is important.
-
•Describe the two driving forces behind the free energy change, and write the expression that shows how they are related to the free energy.
9.7-1. Free Energy
A spontaneous process at constant T and P is one in which
ΔG < 0.
ΔSuniv = ΔS + ΔSsur
qsur = − ΔH,
so ΔSsur = −ΔH/T.
An exothermic reaction releases heat into the surroundings, which increases the entropy of the surroundings. The entropy change in the universe resulting from a reaction carried out at constant temperature and pressure is the following.
ΔSuniv = ΔS − ΔH/T
−TΔSuniv = ΔH − TΔS
-
•ΔG is the change in free energy. It is the energy that is free to do work, i.e., –ΔG = maximum work that can be done by the process at constant T and P. It dictates the spontaneous direction of the reaction. Reactions proceed spontaneously and do work in the direction in whichΔG < 0.
-
•ΔH is the change in potential energy that results from breaking and forming interactions. Reactions that lower the potential energy of the system(ΔH < 0)reduce the free energy, which is favorable.
-
•TΔS is the change in free energy caused by changing the number of ways energy can be dispersed in the system. Processes that increase the entropy of a system(ΔS > 0)are favored because they lower the free energy of the system by TΔS joules.
9.7-2. Enthalpy and Free Energy
Equation 9.6 indicates that processes tend to minimize their potential energy(ΔH < 0).

Figure 9.4: Enthalpy and Free Energy
-
•Endothermic reactions (Figure 9.4a) require energy, so they cannot occur spontaneously unless they can be driven by TΔS energy. Thus,TΔS > ΔHfor a spontaneous endothermic reaction.
-
•Exothermic reactions (Figure 9.4b) release energy, so they can occur spontaneously unless TΔS is so negative that the released energy is not sufficient overcome the entropy term.
9.7-3. Entropy and Free Energy
Equation 9.6ΔG = ΔH − TΔS
indicates that processes tend maximize their entropy (ΔS > 0).

Figure 9.5: Entropy and Free Energy
-
•Processes for whichΔS > 0release energy that can be used to drive reactions uphill in enthalpy, but any TΔS energy that is not used in this manner is free to do work.
-
•Processes for whichΔS < 0require energy, which must be supplied from ΔH if the process is to be spontaneous.
9.7-4. Free Energy and Reaction Spontaneity
There are driving forces in both the forward and reverse directions of most reactions, so double arrows are often used in chemical equations. A B shows that there are driving forces for both A → B and A ← B. ΔG is the difference between these forces. If
B shows that there are driving forces for both A → B and A ← B. ΔG is the difference between these forces. If ΔG < 0,
the forward driving force is greater, so there is a net force in the forward direction and the reaction consumes A to produce B. When ΔG > 0,
the reverse driving force is greater, so there is a net force in the reverse direction, and the reaction produces A by consuming B. If ΔG = 0,
the two forces are equal, and there is no net driving force. At this point there is no longer a change in concentrations as the reaction has reached equilibrium. Indeed, ΔG = 0
is the thermodynamic definition of equilibrium at constant temperature and pressure. As summarized in the Table 9.5, the following is true.
The sign of ΔG indicates the spontaneous direction of reaction.
H2O(g). Initially, only liquid is present, so there is no driving force in the reverse direction (no condensation). Consequently, ΔG < 0
and the liquid begins to evaporate. However, as the pressure of the vapor increases, so does the driving force in the reverse direction, so ΔG gets less negative. Eventually, the pressure of the vapor is such that the driving force in the reverse direction equals that in the forward direction, ΔG = 0,
and the process has reached equilibrium.

Table 9.4: The Sign of the Free Energy and the Direction of Reaction
9.8 Standard Free Energy and the Extent of Reaction
Introduction
The difference between the free energy of a reaction (ΔG) and the standard free energy of reaction (ΔG°) is an important one and one that is often confused by students. In this section, we examine the meaning of these two important thermodynamic properties.Objectives
-
•Use the standard enthalpy and entropy of a reaction to predict the conditions of temperature at which the standard free energy is most likely negative.
9.8-1. Standard Free Energy
As is the case with enthalpies, entropies and free energies of reaction are usually given for the reaction carried out under standard conditions. The standard free energy is defined in terms of the standard enthalpy and entropy of reaction as the following.ΔG° = ΔH° − TΔS°
Exercise 9.13:
Predict the signs of ΔH° and ΔS°. Then use ΔG° = ΔH° – TΔS° to predict the temperature conditions under which ΔG° is likely to be negative.
(a) H2(g) + Cl2(g) → 2 HCl(g) ΔH° = –180 kJ/mol
-
What is the sign of ΔH°?
-
+ ΔH° = –180 kJ/mol
-
–
-
What is the sign of ΔS°?
-
+ The number of moles of gas is the same on each side of the equation.
-
– The number of moles of gas is the same on each side of the equation.
-
~ 0
-
ΔG° is most probably negative at which of the following?
-
all temperatures ΔS° ~ 0, so it has little effect. ΔH° << 0, so ΔG° << 0 and the reaction is extensive at all temperatures.
-
no temperatures ΔS° ~ 0, so it has little effect, and ΔH° << 0.
-
low temperatures only ΔS° ~ 0, so it has little effect, and ΔH° << 0.
-
high temperatures only ΔS° ~ 0, so it has little effect, and ΔH° << 0.
(b) CCl4(l) → CCl4(g)
-
What is the sign of ΔH°?
-
+
-
– Evaporation is always endothermic because the energy of the molecules in the gas is greater than that of those in the liquid.
-
What is the sign of ΔS°?
-
+
-
– The evaporation produces gas.
-
~ 0 The evaporation produces gas.
-
ΔG° is most probably negative at which of the following?
-
all temperatures ΔS° > 0, which is good. ΔH° > 0, which is bad.
-
no temperatures ΔS° > 0, which is good. ΔH° > 0, which is bad.
-
low temperatures only ΔS° > 0, which is good. ΔH° > 0, which is bad.
-
high temperatures only ΔS° > 0, which is good. ΔH° > 0, which is bad. Thus, the reaction is most extensive at high temperatures where the TΔS term can dominate and make ΔG° < 0, which makes the reaction extensive.
(c) P4(s) + 6 Cl2(g) → 4 PCl3(g) ΔH < 0
-
What is the sign of ΔH°?
-
+ The problem states that ΔH° is negative.
-
–
-
What is the sign of ΔS°?
-
+ The reaction consumes gas (six moles go to four moles).
-
– The reaction consumes gas (six moles go to four moles), so ΔS° < 0.
-
~ 0 The reaction consumes gas (six moles go to four moles).
-
ΔG° is most probably negative at which of the following?
-
all temperatures ΔS° < 0, which is bad. ΔH° < 0, which is good. Thus, the TΔS° term must be minimized.
-
no temperatures ΔS° < 0, which is bad. ΔH° < 0, which is good. Thus, the TΔS° term must be minimized.
-
low temperatures only ΔS° < 0, which is bad. ΔH° < 0, which is good. Thus, the TΔS° term must be minimized. At low temperature, the ΔH° term will dominate making ΔG° negative and the reaction extensive.
-
high temperatures only ΔS° < 0, which is bad. ΔH° < 0, which is good. Thus, the TΔS° term must be minimized.
9.8-2. Extent of Reaction
ΔG° is the value of ΔG when all reactants and products are in their standard states. Consider the process:A(g)
B(g). If ΔG° < 0, the reaction is spontaneous in the forward direction when both pressures are 1 atm, so equilibrium is attained by consuming A, which reduces PA to less than 1 atm, and producing B, which increases PB to more than 1 atm. If PB > PA
at equilibrium, the reaction is said to be extensive because the amount of product at equilibrium is much greater than the amount of reactant. If ΔG° > 0, the process is spontaneous in the reverse direction at standard conditions. Equilibrium in this case is attained when PA > PB,
so there is more reactant than product at equilibrium and the reaction A equilibrium B is not extensive. We conclude the following.
ΔG° indicates the extent of reaction.
9.8-3. Extent Exercise
Exercise 9.14:
One mole each of NH3 and N2H2 are allowed to react in identical 1-L containers at 298 K. Which flask would contain the greater number of moles of H2 when the reactions are complete?
You should read the replies to both answers to better understand the role of ΔG°.
You should read the replies to both answers to better understand the role of ΔG°.
-
2 NH3 → N2 + 3 H2 ΔG° = +34 kJ Stoichiometry predicts that 1.5 mol H2 forms for each mole of NH3 that reacts, but ΔG° > 0 for this reaction, so it is not extensive. Consequently, very little ammonia decomposes (less than 1%) at 298 K and very little H2 is produced by this reaction at this temperature.
-
N2H2 → N2 + 2 H2 ΔG° = –160 kJ Stoichiometry predicts that 2 mol of H2 should form for each mole of N2H4 that reacts, but the amount that reacts is dictated by the extent of reaction. Since ΔG° is large and negative, we conclude that the reaction is extensive and that essentially all of the N2H4 does react. Consequently, this flask would contain close to 2 moles of H2 at the end of the reaction.
9.8-4. Extent of Reaction Versus Temperature
ΔG° = ΔH° – TΔS° can be re-arranged to give the common form of a straight line(y = mx + b)
as follows.
| y | = | m | x | + | b |
| ΔG° | = | –ΔS° | T | + | ΔH° |
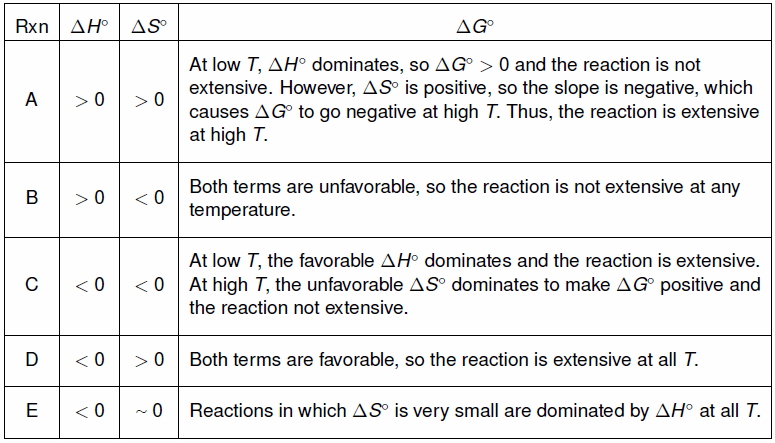
Table 9.5: Standard Free Energy Versus Temperature

Figure 9.6: Standard Free Energy Versus Temperature
9.8-5. Summary
At very low temperatures, the TΔS° term is negligible, and ΔG° has the sign of ΔH°. At very high temperatures, the TΔS° term dominates (unless ΔS° ~ 0), and ΔG° has the sign of –ΔS°.
Table 9.6: Predicting the Extent of Reaction
9.9 Activation Energy
Introduction
Thermodynamics compares the initial and final states only. It does not consider how the transition from one state to another is accomplished; that is the domain of kinetics. Energy considerations are also an important part of kinetics, and we use the one-step displacement of iodide by hydroxide to examine reaction energetics in this section.Objectives
-
•Define reaction coordinate, transition state, and activation energy.
-
•Explain how the enthalpy of reaction is related to the activation energies of the forward and reverse reactions.
9.9-1. The Reaction
In this section we examine both the thermodynamics and kinetics of the following one-step reaction in which iodide is displaced by a hydroxide ion.Thermodynamics is concerned only with differences between the initial and final states of the reaction, while kinetics is concerned with how the reaction proceeds.
Figure 9.7
9.9-2. Thermodynamic View
Thermodynamics is concerned only with the difference between the initial and final states.
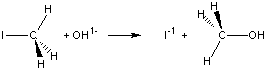
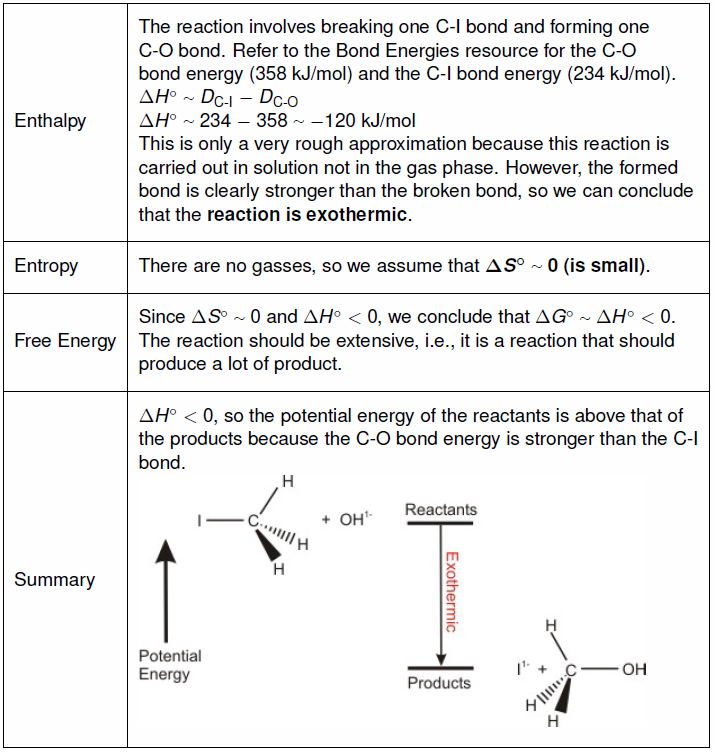
Table 9.7
9.9-3. Kinetic View
Reaction Diagram and Transition State
- Viewing the Video
-
•View the video in this window by selecting the play button.
-
•Use the video controls to view the video in full screen.
-
•View the video in text format by scrolling down.
Kinetics deals with the transition from reactants to products.
Reactants must go through a transition state to reach the products, and the energy required to reach the transition state is called the activation energy.
ΔH = Ea(f) − Ea(r),
so Ea(f) < Ea(r)
for exothermic reactions, but Ea(f) > Ea(r)
for endothermic reactions.

Figure 9.8: Reaction Diagram for CH3I + OH = CH3OH + I1–
9.10 Rates of Reaction and The Rate Law
Introduction
In this section, we examine the factors that dictate the rate of a reaction, and then pull all of the factors together into a rate law for the reaction.Prerequisites
-
•7.1-11. Molarity (Determine the molarity of a substance in a mixture.)
-
•7.1 Gases
Objectives
-
•Express the collision frequency between several particles in terms of their molar concentrations.
-
•State the two factors that dictate the fraction of collisions that lead to the transition state.
-
•Write the rate law that governs a reaction that occurs in one step given the balanced chemical equation for the reaction.
-
•Define the term catalyst and explain how a catalyst functions.
9.10-1. Collision Frequency
The frequency of collisions between two particles is proportional to the product of their concentrations.
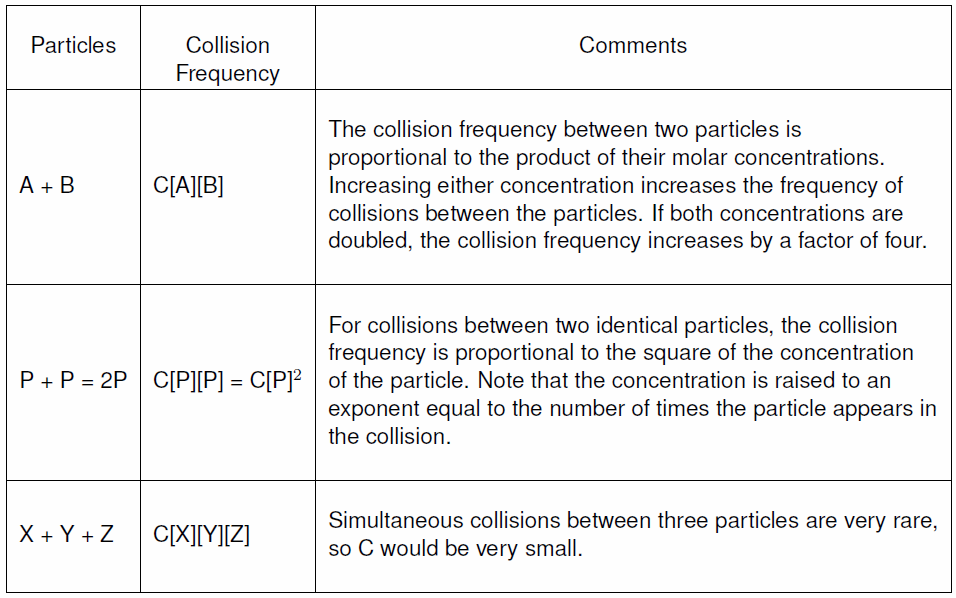
Table 9.8
9.10-2. Orientation Effects
Only a fraction of colliding particles are oriented properly for reaction.

Figure 9.8: Orientation Effect
(a) OH1– ion attacks the iodine atom not the carbon, so transition state cannot form; (b) OH1– ion attacks the correct position, but it is turned so that a C-O bond cannot form; (c) the only orientation that can lead to the transition state.
9.10-3. Energy Effect
Only a fraction of colliding particles have sufficient energy to react.
9.10-4. Rate Law
Many reactions involve several steps to convert the reactants to the products. For example, the reaction between hydrogen and oxygen is a complicated reaction that involves many steps. However, some reactions, such as the displacement reaction between CH3I and OH1–, occur in a single collision (step). Consider the rate of the reaction A + B → products. The rate of a reaction equals the fraction of collisions (X) that lead to the transition state times the collision frequency (F), so we may write the following.rate = XF
F = C[A][B]
rate = XC[A][B] = k[A][B]
The rate law for a simple, one-step reaction is equal to the rate constant for the reaction times the product of the concentrations of the reactants each raised to an exponent equal to their coefficient in the balanced chemical equation.
9.10-5. Rate Law Exercise
Exercise 9.15:
What are the rate laws for the following reactions, which all occur in a single step?
O3 + O → 2 O2
rate =
o_k[O_3][O]_s
rate = k[O3][O]
2 NOCl → 2 NO + Cl2
rate =
o_k[NOCl]^2_s
rate =k[NOCl]2
2 CO + O2 → 2 CO2
rate =
o_k[CO]^2[O_2]_s
rate = k[CO]2[O2]
H2 → 2 H
rate =
o_k[H_2]_s
rate = k[H2]
9.10-6. Catalysts
A catalyst increases the speed of a reaction by lowering its activation energy.
-
•Increasing the temperature. Increasing the temperature increases the rate constant by increasing the thermal energy and the average energy of the collisions. Consequently, the fraction of collisions that reach the transition state increases.
-
•Decreasing the activation energy. Lowering the activation energy increases the rate constant by reducing the energy required to reach the transition state. Consequently, a larger fraction of collisions reaches the transition state.
-
•Assuring the correct orientation of reactant. Forcing the reactants to align correctly when they interact, increases the rate constant because a larger fraction of the interactions result in the transition state.
9.10-7. Ozone Depletion
As an example of catalytic behavior, we consider the problem of ozone depletion in the upper atmosphere. O3 in the upper atmosphere protects us from ultraviolet radiation from the sun. It is removed by reaction by atomic oxygen.O + O3 → 2 O2 Ea = 17 kJ/mol
CF2Cl2 + hv → CF2Cl + Cl
-
1Cl + O3 → O2 + ClOEa' = 2 kJ/mol
-
2ClO + O → Cl + O2Ea'' = 0.4 kJ/mol
(O + O3 → 2 O2).
The chlorine atoms speed the reaction but are unchanged by it, so they are a catalyst for the reaction. They provide a different path with a lower activation energy (the activation energies for the two steps are 2 and 0.4 kJ/mol, respectively), so the reaction is much faster in the presence of chlorine.

Figure 9.9: Reaction Diagram for Ozone Depletion
Shown is the reaction diagram for the ozone depletion reaction in the absence (black curve) and presence (red curve) of chlorine atoms.
9.11 Equilibrium and the Equilibrium Constant
Introduction
Although it is common to say that a reaction has stopped, reactions don't really stop. Instead, they reach a dynamic equilibrium in which the molecules on the right side of the chemical equation continue to produce those on the left and vice versa. At equilibrium, however, the amounts of products and reactants are no longer changing because they are being consumed and produced at the same rate. In this section, we discuss the equilibrium process from the viewpoints of both kinetics and thermodynamics and introduce the equilibrium constant.Objectives
-
•Explain what is meant by a 'dynamic equilibrium.'
-
•Write the equilibrium constant expression for a reaction from the balanced chemical equation.
-
•Determine the value of an equilibrium constant from the equilibrium concentrations (or pressures) of all reactants and products.
-
•Determine the equilibrium concentration (or pressure) of one substance from the equilibrium concentrations (or pressures) of all other substances in the equilibrium mixture and the value of the equilibrium constant.
-
•Explain what is meant by an extensive reaction and predict whether a reaction is extensive from the value of its equilibrium constant.
-
•Relate the equilibrium constant to the standard free energy of reaction.
-
•Use standard enthalpy and entropy changes to predict the extent of reaction.
-
•Predict the effect that a temperature change has on the equilibrium constant of a reaction.
9.11-1. Equilibrium and the Equilibrium Constant
A reaction reaches equilibrium when the rates of the forward and reverse reactions are equal.
) are used in reactions at equilibrium to emphasize that the reaction takes place in both the forward and reverse directions. Consider the reaction: CH3OH1– I1–CH3OH.
| Reaction | Rate | |
| Forward Reaction | CH3I + OH1– → I1– + CH3OH | Rf = kf[CH3I][OH1– ] |
| Reverse Reaction | I1– + CH3OH → CH3I + OH1– | Rf = kf[CH3OH][I1– ] |
At equilibrium,
Rf = Rr,
so we can write the following.
kf[CH3I][OH1−] = kr[CH3OH][I1−]
| kf |
| kr |
| [CH3OH][I1−] |
| [CH3I][OH1−] |
K =
| [CH3OH][I1−] |
| [CH3I][OH1−] |
The equilibrium constant expression equals the equilibrium concentrations of the substances on the right side of the chemical equation (the products) divided by the equilibrium concentrations of the substances on the left side of the chemical equation (the reactants). The concentration of each substance is raised to an exponent equal to the coefficient of the substance in the balanced chemical equation.
9.11-2. Some Examples of Equilibrium Constant Expressions
The concentration of a gas in an equilibrium constant expression equals its partial pressure in atmospheres, while the concentration of a solute is given by its molar concentration. Solids and liquids enter as unity (1).
-
•Pure solids and liquids enter the equilibrium constant expression as one (1) because their concentrations do not change during a reaction.
-
•Gases enter K as their partial pressures in atmospheres. Thus, substance X would enter the expression as PX if X was a gas.
-
•Substances that are dissolved in solution enter K as their molar concentrations. Substance X enters the expression as [X] if X is dissolved in solution.
aA(g) + bB(aq) xX(s) + yY(aq)
xX(s) + yY(aq)K =
=
| 1x[Y]y |
| PaA[B]b |
| [Y]y |
| PaA[B]b |
Table 9.9: Some Examples of Equilibrium Constant Expressions
9.11-3. K Calculation Exercise
Exercise 9.16:
Use the following chemical equation to answer the questions:
N2O4 2 NO2.
2 NO2.
Calculate the value of K at a temperature where the following equilibrium concentrations are found: PNO2 = 0.12 atm and PN2O4 = 0.075 atm.
K =
0.192___
K =
= 0.19
| P2NO2 |
| PN2O4 |
What is the equilibrium pressure of N2O4 if the equilibrium pressure of NO2 is 0.36 atm and K = 0.19?
0.6821___
K =
⇒ | P2NO2 |
| PN2O4 |
PN2O4 =
=
= 0.68 atm
| P2NO2 |
| K |
| 0.362 |
| 0.19 |
9.11-4. Extensive Reactions
An extensive reaction is one in which K >> 1; that is, one in which the equilibrium concentrations of the products exceed those of the reactants.
Exercise 9.17:
Indicate whether the products, reactants, or neither would dominate the equilibrium mixture formed by adding equal amounts of all reactants in each of the following reactions.
-
products
-
reactants K >> 1, so the products (numerator) dominate in the equilibrium mixture. That is, the equilibrium concentration of HI will be much greater than that of H2 or I2.
-
neither K >> 1, so the products (numerator) dominate in the equilibrium mixture. That is, the equilibrium concentration of HI will be much greater than that of H2 or I2.
H2(g) + I2(g) 2 HI(g) K = 600
2 HI(g) K = 600
-
products K << 1, so the reactants (denominator) dominate in the equilibrium mixture. That is, the equilibrium concentration of HF will be much greater than that of F1– or H3O1+.
-
reactants
-
neither K << 1, so the reactants (denominator) dominate in the equilibrium mixture. That is, the equilibrium concentration of HF will be much greater than that of F1– or H3O1+.
HF(aq) + H2O(l) F1−(aq) + H3O1+(aq) K = 7 × 10−4
F1−(aq) + H3O1+(aq) K = 7 × 10−4
-
products K ~ 1, so neither side dominates. That is, the reactants and products will have similar concentrations in the equilibrium mixture.
-
reactants K ~ 1, so neither side dominates. That is, the reactants and products will have similar concentrations in the equilibrium mixture.
-
neither
HCN(aq) + NH3(aq) CN1−(aq) + NH41+(aq) K = 0.7
CN1−(aq) + NH41+(aq) K = 0.7
9.11-5. Thermodynamic Definition of K
Thermodynamic Basis for Equilibrium
There is a driving force for the forward reaction that depends upon the concentrations of the substances on the left side of the chemical equation and one for the reverse direction that depends upon the concentrations of those on the right side. The difference between these two driving forces is ΔG. Thus, there are three possibilities.-
•ΔG < 0:the driving force in the forward direction is greater, so the reaction proceeds from left to right (→).
-
•ΔG > 0:the driving force in the reverse direction is greater, so the reaction proceeds from right to left (←).
-
•ΔG = 0:the driving forces are equal, so the reaction is at equilibrium ().
ΔG = 0
is the criterion for equilibrium, but it is the value of ΔG° that indicates where the equilibrium will be established. Indeed, the equilibrium constant for a reaction is related to the standard free energy of the following reaction.
ΔG° = –RT ln K.
-
•ΔG° ~ 0 → K ~ 1 (ln 1 = 0). When K ~ 1, the numerator and denominator of the equilibrium constant expression are nearly the same, so the equilibrium mixture is composed of comparable concentrations of both the reactants and products.
-
•ΔG° << 0 → K >> 1. K >> 1 when the numerator (product concentrations) is much larger than the denominator (reactant concentrations), so the equilibrium mixture is dominated by the products, i.e., the reaction is extensive.
-
•ΔG° >> 0 → K << 1. K << 1 when the numerator (product concentrations) is much smaller than the denominator (reactant concentrations), so the equilibrium mixture is dominated by the reactants, i.e., very little reaction takes place.
9.11-6. Predicting Extent of Reactions
The extent of reaction is determined by ΔG°, but ΔG° is related to the standard enthalpy and entropy changes by the following.ΔG° = ΔH° – TΔS°

Table 9.10
9.11-7. Predicting Extent of Reactions Exercise
Exercise 9.18:
N2(g) + 3 H2(g) 2 NH3(g);
Equilibrium: [N2] ~ [H2] << [NH3]
Use the chemical equation and the equilibrium composition to determine the sign of ΔG°. Use the change in moles of gas to determine the sign of ΔS°. Determine the sign of ΔH° from the other two.
2 HCl(g) H2(g) + Cl2(g); Equilibrium: [H2] = [Cl2] << [HCl]
H2(g) + Cl2(g); Equilibrium: [H2] = [Cl2] << [HCl]-
ΔG°
-
+The concentrations of the products are much less than that of the reactants, so this reaction is not extensive at this temperature. Consequently, ΔG° is positive.
-
−The concentrations of the products are much less than that of the reactants, so this reaction is not extensive at this temperature.
-
~ 0 The concentrations of reactants and products are very different, so ΔG° cannot be close to zero.
-
ΔS°
-
+The balanced equation does not indicate that the number of moles of gas on the product side is greater than on the reactant side.
-
−The balanced equation does not indicate that the number of moles of gas on the product side is less than on the reactant side.
-
~ 0 The balanced equation indicates that the number of moles of gas is the same on both sides of the equations, so ΔS° is small.
-
ΔH°
-
+ΔG° is positive and ΔS° is very small, so ΔH° must also be positive.
-
−
-
~ 0
HF(aq) + NO21–(aq) F1–(aq) + HNO2(aq);
Equilibrium: [F1–] ~ [HNO2] ~ [HF] ~ [NO21–]
F1–(aq) + HNO2(aq);
Equilibrium: [F1–] ~ [HNO2] ~ [HF] ~ [NO21–]
-
ΔG°
-
+The concentrations of the reactants and products are very similar, so ΔG° cannot be a large positive number.
-
−The concentrations of the reactants and products are very similar, so this is not an extensive reaction.
-
~ 0 All concentrations are similar, so K ~ 1 and ΔG° must be close to zero.
-
ΔS°
-
+There are no gases involved, so entropy changes are not expected to be large.
-
−There are no gases involved, so entropy changes are not expected to be large.
-
~ 0 There are no gases involved, so entropy changes are not expected to be large. Thus, ΔS° ~ 0.
-
ΔH°
-
+ΔG° ~ 0 and ΔS° ~ 0, so ΔH° must also be small.
-
−
-
~ 0 ΔG° ~ 0 and ΔS
N2(g) + 3 H2(g)
2 NH3(g);
Equilibrium: [N2] ~ [H2] << [NH3]
-
ΔG°
-
+The concentration of the product is much greater than those of the reactants, so ΔG° cannot be large and positive.
-
−The concentration of the product is much greater than those of the reactants, so ΔG° is negative for this reaction at this temperature.
-
~ 0 The concentration of the product is much greater than those of the reactants, so ΔG° cannot be very small.
-
ΔS°
-
+Gas is consumed by the reaction, so ΔS° cannot be positive.
-
−Two moles of gas are formed for every four that react. Gas is consumed by the reaction, so ΔS° is negative.
-
~ 0 Gas is consumed by the reaction, so ΔS° is not small.
-
ΔH°
-
+
-
−ΔG° < 0 and ΔS° < 0, so ΔH° must also be negative.
-
~ 0
9.11-8. Effect of T on K
The value of an equilibrium constant varies with temperature. Changes in temperature change the thermal energy of the molecules, which changes the fraction of molecules at higher energy. The fraction of molecules at higher energy increases when the temperature is increased.-
•Exothermic reactions: The reactants are on the higher energy side, so increasing the temperature increases the fraction of reactant molecules at equilibrium. Consequently, the equilibrium constants of exothermic reactions decrease when the temperature is increased.
-
•Endothermic reactions: The products are at higher energy, so increasing the temperature increases the fraction of product molecules at equilibrium. Consequently, the equilibrium constants of endothermic reactions increase when the temperature is increased.
9.11-9. Example
Exercise 9.19:
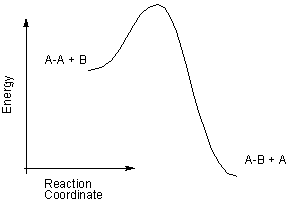
-
The equilibrium concentration of A-B is greater at which of the following?
-
high T
-
low T A-B is a product, which is at lower energy in exothermic reaction. Substances are low energy are favored at low T. Lowering the temperature increases the concentrations of the reactants and decreases the concentrations of the products, which has the effect of decreasing the value of the equilibrium constant. This reaction becomes less extensive as the temperature increases because it is exothermic.
-
Which is the stronger bond?
-
A-A
-
A-B ΔH < 0, but ΔH ~ (A-A bond energy) - (A-B bond energy). Consequently, A-B bond energy is greater than the A-A bond energy.
-
Is the reaction extensive?
-
Yes ΔH° < 0, and the number of gaseous molecules is unchanged during the reaction, so ΔS° ~ 0. Consequently, ΔG° < 0 and the reaction is extensive.
-
No
-
Which reaction has the higher rate constant?
-
forward If the orientation factor is assumed to be similar for the two reactions, the one with the lower activation energy has the larger rate constant. The activation energy for the forward reaction (A-A + B → A-B + A) is less than that for the reverse reaction (A-B + A → A-A + B), so the rate constant for forward reaction is larger.
-
reverse
9.12 Le Châtelier's Principle
Introduction
An open door that is not moving is at equilibrium. However, the equilibrium position of the door can be changed by applying pressure to either side. Reactions at equilibrium react in a similar fashion when they are exposed to a chemical stress.Objectives
-
•Use Le Châtelier's Principle to determine the effect of changing the temperature or the concentration of one substance on the concentration of another substance in the equilibrium mixture.
9.12-1. Le Châtelier's Principle
Equilibrium mixtures respond to the addition or removal of material in a manner consistent with the following principle.-
•Le Châtelier's Principle: a system at equilibrium will respond to a stress in such a way as to minimize the effect of the stress.
-
1Increasing the concentration (or pressure) of one substance in an equilibrium mixture results in some reaction of that substance. The concentrations of the other substances on the same side of the equilibrium are reduced and the concentrations of those on the opposite side are increased by the reaction.
-
2Decreasing the concentration (or pressure) of one substance in an equilibrium mixture results in some reaction of the other substances to replace some of what was removed. The concentrations of the other substances on the same side of the equilibrium as the removed substance all increase, while those of the substances on the opposite side decrease as a result of this reaction.
Exercise 9.20:
Consider the following equilibrium.
Indicate whether each of the following actions would increase, decrease, or not affect the equilibrium concentration of Cl1–
AgCl(s) + 2 NH3(aq) Ag(NH3)21+(aq) + Cl1−(aq) ΔH < 0
Ag(NH3)21+(aq) + Cl1−(aq) ΔH < 0-
adding Ag(NH3)21+
-
increase
-
decrease Equilibrium must shift so as to remove some of the additional Ag(NH3)21+ by reacting it with Cl1–. Therefore, the concentration of Cl1– will decrease.
-
not affect
-
removing Ag(NH3)21+
-
increase Equilibrium must shift so as to replace some of the lost Ag(NH3)21+ by reacting more AgCl and NH3, which increases the concentration of Cl1–.
-
decrease
-
not affect
-
adding solid AgCl
-
increase
-
decrease
-
not affect Adding substances that appears as pure solids or liquids in the equilibrium reaction does not change their concentrations, so it has no effect on the other concentrations.
-
adding NH3
-
increase The equilibrium removes some of the additional ammonia by reacting it with AgCl to produce more Ag(NH3)21+(aq) and Cl1–. Consequently, the equilibrium concentration of Cl1– will increase.
-
decrease
-
not affect
-
increasing temperature
-
increase
-
decrease Changing the temperature is the only change that results in a change of K. Recall that an increase in temperature increases the concentrations of those substances at higher energy, which are the reactants (substances on the left of the chemical equation) in an exothermic reaction (ΔH < 0). Thus, the concentration of chloride ion, which is on the right side of the equation and at lower energy, decreases as the temperature increases.
Alternatively, we can use LeChâtelier's principle by realizing that increasing the temperature is the same as adding heat, which causes the substances to react so as to remove some of the heat. Heat is a product of an exothermic reaction, so the concentrations of all products (substances on the right side of the equation) decrease, while the concentrations of the reactants all increase. Consequently, the concentration of chloride ion (a product) decreases. -
not affect