How to Use Basic MarvinSketch Functions
Table of Contents
A new, blank molecule is created when you first launch MarvinSketch. You
can immediately begin working with this molecule. A MarvinSketch window can
hold only one molecule at a time, so all work you do within the
canvas is considered part of the same molecule. You can create a new,
blank molecule at any time during your session by choosing
File > New > Clear Desk from the menu bar. This
will clear the desk and discard any unsaved changes to the molecule you were
previously working with (but you can get it back using the Undo option).
The application allows you to work with
multiple molecules in multiple windows by choosing File
> New > New Window.
You can open existing molecule files
(from supported file formats) by
choosing File > Open on the menu bar.
It will load the content of the molecule file into Marvin and discard any
unsaved changes.
Tick the Show preview checkbox to see the contents of the file (molecules,
reactions, queries). A single item is displayed in the preview window,
the text field at the bottom shows the index of the current structure
and the number of structures in the file. When a multiple structure file is selected
(e.g. sdf), the navigation buttons become active. Their functions are: go to first,
go to previous, go to next, go to last. Note, that this is not the selection which structure
to open, it allows you only to check whether this is the file you would like to open.
You can also Paste existing structures from other windows to
the open molecule, as well as Drag&Drop a molecule file into the Marvin
window. Both actions will add the new structure to the currently open
molecule without overwriting the content of the canvas.
You can save the molecule to any of the supported
file formats.
This will allow you to open and work with this molecule later. The
default behavior of the Save
button is to save the molecule to the same file it was opened from,
in the same format. If you want to change the file name or format,
choose Save As.
If you are working with a new molecule, Save
will function the same as Save As.
The Save As Image choice
in the File menu allows you to save an image of the
molecule in the sketcher. Marvin supports the following image
formats: JPEG, PNG, PPM, POVRay, and SVG. Please note that the saved image
cannot be edited in Marvin.
You can print an image of the current molecule by choosing
File > Print. If you print from a single page
document, if the size of your molecule(s) is bigger then the paper size, it will
be shrunken to one page.
If you want to print your molecule(s) to multiple
pages, you have to change the document type to Multipage Document in
File > Document Settings..., and arrange the
structures on the pages.
Multipage molecular documents help to work with large drawings by dividing them into pages.
You can create a multipage molecular document by choosing
File > Document Settings..., then checking in the
Multipage document checkbox.
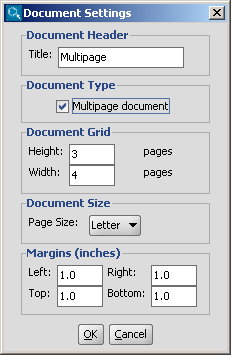
You can set the number of horizontal and vertical pages in the
Document Grid part, and you can also define the title,
the page size and the margins in the corresponding sections of this dialog window.
After pushing the OK button, the following controls become automatically available:
- The items in the View > Pages menu are enabled
- A navigation status bar appears on the bottom of the window
- The frame of the pages appear on the canvas, while the title, the margins and
the page numbers are displayed on each page
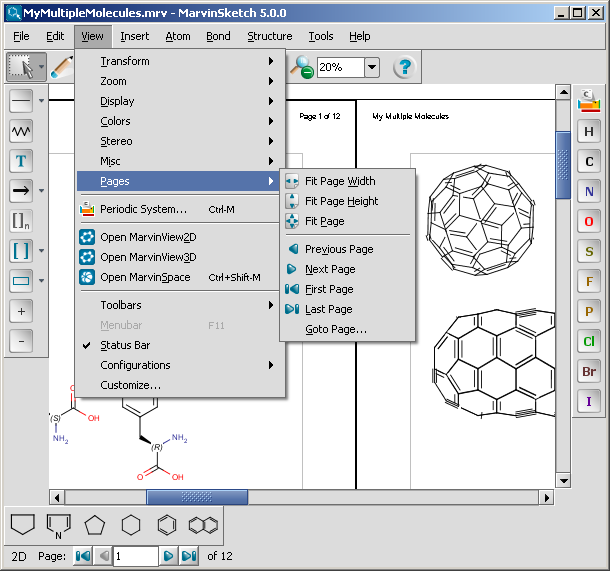
The navigation status bar and the items in the View > Pages menu are
available only if the Multipage document checkbox is set.
The status bar contains information about
the current page number in a text field and the number of all pages on a label.
It also contains a collection of buttons to
aid your quick navigation in the document. You can go the first, previous, next, and last page
using them. Alternatively, you can go directly to a specific page
by entering a number in the current page field and pressing enter.
All the navigation possibilities: go to first, previous, next, last, specific
pages are available from the View > Pages menu as
well. In addition, some automatic page zooming functions are also available in this menu, such as:
- View > Pages > Fit page height adjusts the
height of the current page to the height of the canvas.
- View > Pages > Fit page width adjusts the
width of the current page to the width of the canvas.
- View > Pages > Fit page adjusts the height
and/or the width of the current page to see the whole page, and places it
centralized within the canvas.
You can create structures using atoms, bonds, and templates.
- Select an atom from the Atoms Toolbar,
the Periodic Table dialog window, or by
shortcut.
- Move the mouse into the molecule
canvas. You will see the symbol of the selected item at the tip of
your cursor. It can be placed in the molecule by left-clicking on
the desired location.
- Marvin is chemically intelligent.
It will account for implicit hydrogens and set the charge according
to valence rules.
- You can replace any atom in the molecule by placing a new
atom on top of it.
- Select a bond type using the
Bond toolbar button or by
shortcut.
- To link two existing atoms, click
on one then drag the cursor to the other.
- Marvin will allow you to draw a
bond between any two atoms in the molecule. Valence errors
will be highlighted (if that option is enabled).
- To draw a bond from a single atom,
simply click the atom. A carbon atom will be added at the other end
of the bond.
- If you add a bond to empty canvas
space, a carbon atom will be added to each end of the bond.
- You can replace any bond in the
molecule by placing a new bond type on top of it.
- Bond types can also be changed using the
Bond pop-up menu. Single
bonds can be changed to Double or Triple by
left-clicking on them.
MarvinSketch provides several predefined chemical structures,
called templates or structure templates (e.g., cyclopenthane, cyclohexane, benzene).
They are categorized to template groups like Amino Acids, Polycyclics, etc.
The following template groups are available in MarvinSketch by default:
- Generic
- Rings
- Amino Acids
- Bicyclics
- Bridged Polycyclics
- Crown Ethers
- Cycloalkanes
- Hetero Cycles
- Polycyclics
- Configuration1
- My Templates
The templates can be accessed via the Advanced Templates Toolbar
or through the Insert > Template Library menu.
Steps of drawing with templates:
- Select a template using the
Template Library or the Advanced Templates Toolbar area.
- In case the template structure contains any S-groups, the group(s)
can be optionally expanded or contracted by pressing the Shift button.
- Place the template structure by left-clicking on the
desired location.
You can use coordinate bond to represent coordination compounds (ferrocenes, metallocenes).
For example:
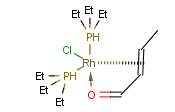
The coordinate bond type can represent the connection between an atom and a group of atoms.
The coordinate bond has two kinds of appearance according to IUPAC recommendation:
- arrow between two atoms,
- dashed line between an atom and a group of atoms.
To draw a bond between two atoms just choose the coordinate bond from
the bond list and draw the bond by specifying the required direction. To draw a
bond between an atom and a group of atoms you need to create
a multi-center attachment point to represent the group of atoms.
To draw a coordinate bond between an atom and a group of atoms
- Select the atoms to be represented at one end of the coordinate bond by a multi-center.
- Choose "Structure/Add/Multi-center" from the main menu or
"Add/Multi-center" from the contextual menu. A multi-center represented by a "*" will
be added. If you move the cursor to the multi-center the represented atoms are
highlighted (blue circle around the atom labels).
- Draw a coordinate bond from the multi-center and edit the other end of the bond if required.
The "*" representing the multi-center disappears after bond drawing.
- Repeat steps 2-4 to draw further multi-centers and coordinate bonds if required.
A Markush structure is a description of compound classes by generic notations.
They are often used for patent claims and for combinatorial libraries.
Link R-groups, link nodes, atom lists, position variation and
repeating units with repetition ranges are commonly used features in the representation of Markush structures.
Position variation (Variable point of attachment)
You can create a variable point of attachment to represent a variable connection point to a group of atoms.
The representation is similar to the above mentioned multi-center bonds.
For example:
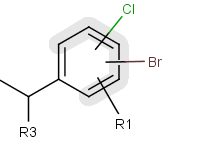
The alternative attachment points are displayed with grey shadow.
If you move the cursor to the center (the bond ending in the ring) the represented atoms are
highlighted (blue circle around the atom labels).
To draw a position variation
- Draw the structure that will include the position variation.
- Select the alternative connection point atoms.
- Choose "Structure/Add/Multi-center" from the main menu or
"Add/Multi-center" from the contextual menu. A multi-center represented by a "*" will
be added. If you move the cursor to the multi-center the represented atoms are
highlighted (blue circle around the atom labels).
- Draw a bond from the center and edit the bond if required. The represented atoms
are displayed with grey shadow after this step. The "*" representing the multi-center
disappears after bond drawing.
- Repeat step 2-4 to draw further variable points if required.
Frequency variation (repeating unit with repetition ranges)
A sequence of ranges to specify the repetition can also be used in a special
group called repeating unit with repetition ranges. For example:
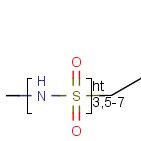
Here the repetition range is "3,5-7". The repetition count for the included structure
(enclosed by the brackets) can be: 3,5,6 or 7.
See Repeating units with repetition ranges
for further information on drawing this feature.
You might need to flip the whole or parts of the structures. These operations
are located in the Edit > Transform menu. If no selection is made, the operation
will be executed on the whole structure (except for Group Flip). The flip operation is equal
to a rotation of 180° around a horizontal or vertical axis in the plane of
the drawing. All flips result in stereocenter retention.
| Horizontal flip |
 |
| Vertical flip |
 |
Horizontal Flip, Vertical Flip
Flip a selection:
- Select part of the structure.
- Right-click on the structure or go to Edit > Transform menu.
- Click on the command.
Flip the whole structure without selection:
- Go to the Edit > Transform menu.
- Click on the command.
Note: If no structure is selected, the right-click on the canvas will not offer the
flip command.
Group Flip
The Group Flip operation can be executed only on a selected structure connected
to the rest of the molecule by only one bond (of any type): the selection
can not be in the 'middle' of a molecule. The selection is not permitted for disjunctive structures either.
The Group Flip operation rotates the selected group by 180° around an axis
set on the bond connecting the selection to the rest of the molecule. Stereocenters
in the molecules are retained, the wedge bond styles change to keep the stereo information.
| Group flip |
 |
- Select part of the molecule.
- Right-click on the canvas and select Transform > Group Flip; or go to
the Edit > Transform menu.
- Select Flip > Group.
Apart from flipping Marvin is able to produce mirror images of the molecules or parts of.
These operations can be found in the Edit > Transform menu. If no selection
is made, the operation will be executed on the whole structure. Stereocenters will be
inversed. Mirroring horizontally means that the theoretical mirror is vertical and
placed perpendicular to the canvas (left-to-right mirroring); the vertical
mirroring means the mirror is horizontal and perpendicular to the canvas
(upside-down mirroring).
| Horizontal mirroring |
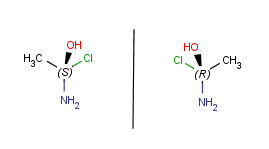 |
| Vertical mirroring |
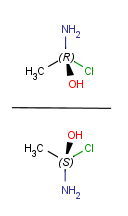 |
Mirror a selection:
- Select part the molecule.
- Right-click on the canvas or go to the Edit > Transform menu.
- Click the command.
Mirror the whole structure without selection:
- Go to the Edit > Transform menu.
- Click on the command.
You can place a reaction arrow on the canvas at any time, even on
a blank canvas. Only one reaction is allowed per molecule.
- Select the
React
button. You will see the reaction arrow on the tip of the cursor when
you move the mouse into the canvas area.
- Click the location of the tail of
the arrow.
- Drag the mouse and release at the location of the head.
Once you have placed a reaction arrow
on the canvas, MarvinSketch considers each part of the molecule in
relation to the reaction.
All parts of the molecule that are before the arrow are considered reactants.
Every molecule after the arrow is a product, and the ones placed along the
arrow are considered agents.
The arrow tool provides the easiest manual way to map corresponding reactant
and product atoms. Select the arrow tool, hold down the left mouse button on a
reactant atom, and drag it to the corresponding product atom. The same map
number is added to both atoms marking, that they represent the same atom on the
two sides of the reaction scheme. There are also keyboard shortcuts for
mapping. Type m8, for example, and click on an atom. Atom map 8 is assigned to
that atom.
Marvin contains an automapper tool as well (available as
Structure > Reaction > Map Atoms) assigning map numbers to all selected atoms of
a reaction automatically.
Map numbers of the selected atoms can be removed by the
Structure > Reaction > Unmap Atoms menu item, or by typing m0 for the selected
atoms.
An R-group query describes a set of derivatives in one query structure (substitution
variation). It can be drawn the following way: First
draw the root structure and place some R atoms either from the Periodic System dialog window,
from the popup menu or by typing a corresponding label such as "R1" on the
keyboard.
Then draw the variable R-group ligands and select those substituting the R1 atom.
If you type "R1" now, the selected groups will be marked with "R1". Additional
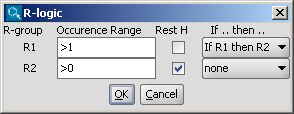
R-group conditions (Occurrence, RestH, If-then) can be set in the R-logic dialog
window available from the Structure > Attributes menu.
To draw the attachment points for the R-definitions, you can use menu
"Group -> Attachment point" from the atom popup menu, or alternatively,
when you draw the R-definitions and the mouse cursor still shows "R1",
clicking on an atom of the definition will toggle the attachment point on that
atom. (Please note that divalent R-groups must have two attachment points
defined.)
Step by step example
- Draw the root structure first.
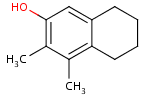
- Move the cursor to the atom where you would like to place the R-group.
(In this example, we place R-groups in place of the terminal carbon atoms.)
When the atom is highlighted (blue circle around the atom label), type the
shortcut of the required R-group ID (e.g. R1). Alternative solution is
selecting the ID from R-group sub-menu of the popup menu by pressing right
mouse button over the atom.
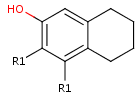
- Draw an alternative ligand with an R-group connection: Move the cursor to an empty place on the canvas (take care that nothing is selected) then press
the shortcut of the next R-group (R2). The "in hand" object changes to the ID of the R-group (R2). (In this example, we add a ligand to terminal oxygen atom.)
Move the cursor over the atom (the terminal oxygen) where you would like to add the new ligand and push the left mouse button. Then drag the mouse. You will
see that the new bond is displayed and its orientation follows the cursor.
Release the mouse button when the bond stands in the right direction.
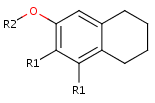
- Draw new fragments to the canvas (separately from the root structure), which
will be the R-group definitions. (In this example, we draw the fragments for
the first R-group definition to the right side and the second R-group
definition will be placed below the root structure.)
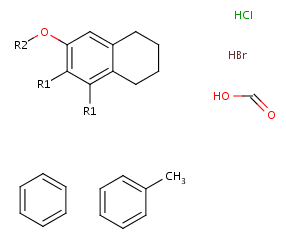
- Next, define the R-groups definitions. To do this, select those fragments
that the first R-group should contain (on the right side).
After the selection, press the shortcut of the R-group ID (R1). The ID and equal sign (R1=) will display beside the selected set and the "in hand" object
will be the R-group ID.
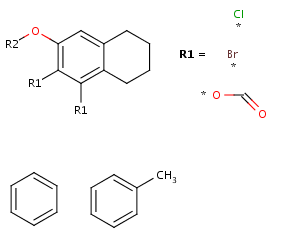
- Define attachment point to R-group members: Click the left mouse button on
atoms where you would like place the attachment points.
Repeat this operation on the other definitions of the R-group.
(In this example, at the third definition, we select the left oxygen atom
for attachment.) Alternatively, you can define R-group attachment points
via the popup menu (by selecting Attachment point option from the Group
menu on an atom of an R-group definition.
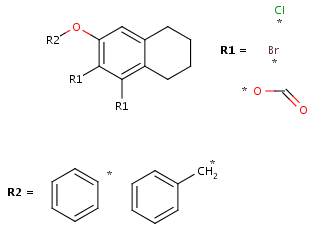
- Create the second R-group by repeating the last two steps on the two
remaining fragments.
- You can define additional conditions, such as occurrence, rest H and if-then
expressions to R-groups in the R-logic dialog window. To do this,
select menu option Structure > Attributes -> R-logic.
After setting the conditions in the R-logic dialog window, press the
OK button to apply the changes.
Abbreviated S-groups are used to represent a part of a structure with a text abbreviation.
- Insert an abbreviated group into your sketch: type the name of the abbreviation, to complete a longer name,
press ENTER or END after typing the first few characters. If the cursor was placed
over an atom, it will be automatically changed to the abbreviated group. If no atom was selected,
the abbreviation is placed on the cursor. Click on the canvas to place it. If you would
like to ungroup an S-group before placing it to the canvas,
press the SHIFT button before you release the mouse on the desired
location.
- Create an abbreviated S-group: select the structure to abbreviate then open
the Create Group dialog window from the Structure > Group submenu. Name the group
in the appearing dialog window.
You can retrieve the hidden structure from the text abbreviation with the "Expand"
function and hide the structure with the "Contract" function.
Manipulation with abbreviated S-groups is possible with "Expand", "Contract",
"Ungroup", and "Remove" from the Group submenu.
A short animation about abbreviated groups:
Expand and ungroup abbreviated groups.
After creating the S-group (see previous section), right-click the corresponding
atom and click attachment point. This way you define the connection point of this
group. Then select the group, right-click and press Add to My Templates. The template
can be inserted by typing its name and clicking on the canvas.
Syntax of the abbreviated S-group name
Numbers are automatically subscripted unless "\n" is used or at the start of string.
Charges (+, -, ++, --, 3+ etc.) are automatically superscripted at end of string
or if the following character is a closing parenthesis.
Allowed control sequences in the abbreviated S-group name:
- \s - subscript
- \S - superscript
- \n - normal mode.
Example: \S13CH4
Multiple S-groups are used to represent a repeating part in a structure with a shorter
form. To create a multiple S-group first select the structure to multiply then open
the Create Group dialog window from the Structure > Group submenu.
Here you can specify a positive repeating count depending on how many times you want the structure to be repeated.
You can retrieve the whole structure from the condensed form with the "Expand"
function and shorten the structure with the "Contract" function.
Manipulation with multiple S-groups is possible with "Expand", "Contract",
"Ungroup", and "Remove" from the Group submenu.
For example:
These features can be expressed by brackets (S-groups) of type component,
unordered mixture (also called mixture)
and ordered mixture (also called formulation).
A component here is a set of atoms contained by a component bracket.
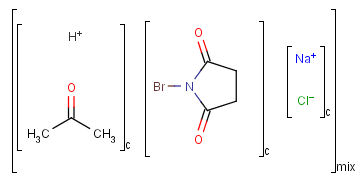
Ordered and unordered mixtures
An unordered mixture (denoted by "mix" at the bottom of the right bracket)
consists of several unordered components (denoted by "c" at the bottom of the right bracket).
For these types of mixtures, the order of addition during the
preparation is not important. Example:
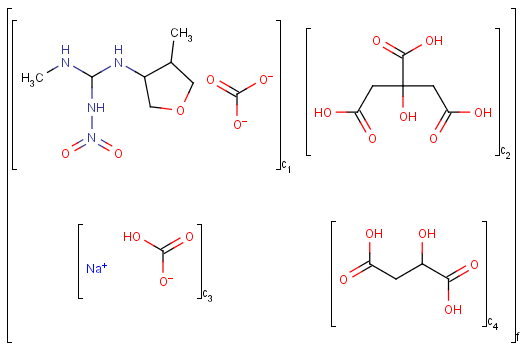
Ordered mixtures, on the other hand contain ordered
components, which define the order of addition. Example:
To draw an unordered component
- Draw the structures that form the mixture.
- To define a structure as a component in a mixture, first select the structure.
- Choose "Group..." from the contextual menu (right mouse click on the selected structure).
- In the "Create Group" dialog window choose "Component (c)" from the "Type" list.
- The "Order" field should be empty or should contain "none".
If the "Order" field already contains a number, just
delete it (you can type in "none" as well).
- Click OK.
To draw an ordered component
- Draw the structures that form the mixture.
- To define a structure as a component in a mixture, first select the structure.
- Choose "Group..." from the contextual menu (right mouse click on the selected structure).
- In the "Create Group" dialog window choose "Component (c)" from the "Type" list.
- If this is the first component of the mixture,
click the "Order" field and enter "1" in place of "none".
If the "Order" field already contains a number Marvin will automatically increment the
"Order" field for subsequent components.
- Click OK.
To draw a mixture
- Create and select the components to form the mixture.
- Choose "Group..." from the contextual menu
(right mouse click on the selected components).
- In the "Create Group" dialog window choose the type
("Ordered mixture(f)" or "Unordered mixture(for)") from the "Type" combobox.
- Click OK.
To change the type of a mixture
- Select the mixture to change the type.
- Choose "Ungroup" from the contextual menu
(right mouse click on the selected mixture).
- Change the type of the included components.
- Select the components to create the new mixture.
- Create the new mixture from the selected components.
To add a new component to a mixture
- Select the mixture to add the new component.
- Choose "Ungroup" from the contextual menu
(right mouse click on the selected mixture).
- Create the new component to add.
- Select the new and old components and create a mixture again.
To delete a component from a mixture
- Select the mixture to delete the component from.
- Choose "Ungroup" from the contextual menu
(right mouse click on the selected mixture).
- Delete the component.
- Select the remaining components and create a mixture again.
The polymer structure consists of structural fragments. These fragments are enclosed by polymer
brackets. The meaning of a polymer bracket is that the fragment within the brackets can
repeat with itself. The fragment within the bracket is called repeating unit.
Polymers can be represented as structure-based or source-based polymers depending on how
much structural detail is known.
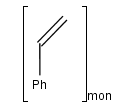
Source-based representation of polymers
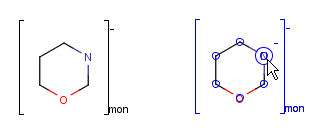
You can use the monomer (mon) or mer (mer) repeating unit types to draw a polymer where only the
source-based representation is known. For example:
To draw a repeating unit select the atoms you want to be included and choose Create Group from Structure menu
or Group/Create Group from contextual menu.
Structure-based representation of polymers
You can use the structural repeating unit type (SRU) to draw a polymer where the structure-based
representation is known.
Each SRU S-group has two or more dedicated bonds, called
crossing bonds, which cross the brackets. The crossing bonds of an SRU show
how the repeating units may connect in several ways to each other within the polymer.
Depending on the number of crossing
bonds and brackets we differentiate the following polymers and connectivities within the polymer:
- Polymers with two crossing bonds.
If the polymer has one crossing bond on each bracket of the SRU
there are three possibilities for the repeating pattern:
- head-to-tail
- head-to-head
- either/unknown
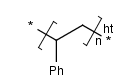
- Ladder-Type Polymers.
Polymers with paired brackets and with two crossing bonds on each bracket are
called ladder-type polymers. Here it must be specified how the two crossing bonds
on each bracket connect to the corresponding bonds of the adjoining repeating units.
Additionally to the head-to-tail, head-to-head connectivity information
there is flip information to specify whether the repeating unit flips around the polymer
backbone when it connects to the adjoining SRU.
These types of information are handled only in case of brackets
with exactly two crossing bonds on both side (head and tail side).
We differentiate the following polymer connectivities:
- head-to-tail with no flip
- head-to-tail with flip
- head-to-head with no flip
- head-to-head with flip
- either/unknown
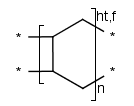
- Polymers with three or more brackets.
If the polymer has three or more bonds with a separated bracket on each bond,
the polymer always has the either/unknown repeating pattern.
The end groups of polymers are often unknown or unspecified which are represented by star atoms (*).
The modified (mod), grafted (grf) and crossliked (xl) form of a structural repeating unit can be drawn as well.
Copolymers
If the structure consists of more than one repeating unit (mon, mer) or structural repeating unit,
Copolymer (co) brackets/groups can be used to represent the structure. Copolymers might contain crossing bonds and
star atoms.
The following copolymers can be drawn:
- random(ran)
- alternating(alt)
- block with or without junction unit (blk)
- copolymer to represent modified polymers (mod)
- copolymer to represent grafted polymers (grf)
- copolymer to represent cross-linked polymers (xl)
For example:
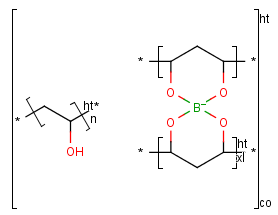
To draw a simple polymer
- Draw the structure that forms the polymer.
- To define a structure as a polymer, first select the structure. Leave out the atoms that should be replaced by "*" (star atoms).
- Choose "Group..." from the contextual menu (right mouse click on the selected structure).
- In the "Create Group" dialog window choose the appropriate type from the "Type" list.
- Set the polymer repeat pattern if necessary.
- Click OK. The star atoms ("*") will be added automatically.
To draw a ladder-type polymer
- Draw the structure that forms the polymer.
- To define a structure as a polymer, first select the structure. Leave out the atoms that should be replaced by "*" (star atoms).
- Choose "Group..." from the contextual menu (right mouse click on the selected structure).
- In the "Create Group" dialog window choose the "SRU polymer" type from the "Type" list.
- Set the polymer repeat pattern if necessary.
- Click OK. The star atoms ("*") will be added automatically.
- To create a bracket that crosses two bonds select the two bonds (they should be already crossed by a single bracket each) and choose
"Groups/Merge Brackets" from the contextual menu.
To draw a copolymer
- Create and select the components to form the copolymer.
- Choose "Group..." from the contextual menu
(right mouse click on the selected components).
- In the "Create Group" dialog window choose the type
("Copolymer (co)", "Copolymer, alternating (alt)", "Copolymer, block (blk)" or "Copolymer, statistical (stat)") from the "Type" list.
- Click OK.
To change the type of a polymer
- Select the polymer to change the type.
- Choose "Ungroup" from the contextual menu
(right mouse click on the selected polymer).
- Create the new polymer from the selected structure(s).
To add a new subpolymer to a copolymer
- Select the copolymer to add the new subpolymer.
- Choose "Ungroup" from the contextual menu
(right mouse click on the selected copolymer).
- Create the new subgroup to add.
- Select the new and old subpolymers and create a copolymer.
To delete a subgroup from a copolymer
- Select the copolymer to delete the subpolymer from.
- Choose "Ungroup" from the contextual menu
(right mouse click on the selected copolymer).
- Delete the subpolymer.
- Select the remaining subpolymers and create a polymer again.
A sequence of ranges to specify the repetition can also be used in a special group called repeating unit with repetition ranges. For example:
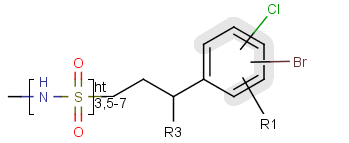
Here the repetition range is "3,5-7". The repetition count for the included structure can be: 3,5,6 or 7.
Syntax of the repetition ranges
The repetition ranges consist of ranges separated by commas. A range can be either a simple non-negative number (e.g. 3)
or two non-negative numbers separated by "-" (e.g. 5-7).
To draw a repeating unit with repetition ranges
- Draw the structure that forms or contains the repeating unit.
- To define a part of the structure as a repeating unit, first select the part of the structure expected to repeat.
- Choose "Group..." from the contextual menu (right mouse click on the selected part).
- In the "Create Group" dialog window choose the type "Repeating unit with repetition ranges" from the "Type" list.
- Set the repetition ranges.
- Click OK.
Four types of groups can be assigned a charge sign: generic, component, monomer and mer groups.
During group creation, you have the option to display the charge on the charged
atom itself or the whole group. In the latter case, the charge will be displayed
outside of the bracket on the right. If any additional charges are added (negative
or positive) the net charge will be calculated and displayed. The charge-bearing
atom can be revealed by pointing the cursor over the group (in select mode). To
replace the charge, select the group and go to the Structure menu, Group submenu and click
Edit Group (or right-click the selected group, and select Edit Group).
To draw a (poly)line, rectangle or text box, use the
Insert menu or the toolbar (if visible). These objects
are depicted in blue color outlines to indicate that any object here does not
bear any chemical meaning like reaction arrows or S-group brackets (in black).
Point the mouse to the
desired position on the canvas, click and hold the left button, move the mouse
and release the button. After placing a text box,
you can immediately use the keyboard to type a text.
To change the contents of a text box, choose
Select mode, click on the box, then use the
keyboard.
The shape of an object is changeable or resizable by dragging one of its points to do it.
To insert the IUPAC Name text box choose it from the Insert
menu, it will be automatically inserted under the structure. The name will be updated
real-time.
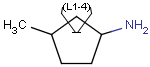
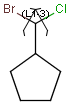
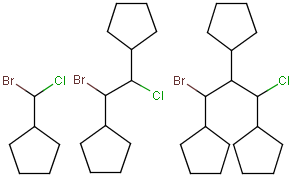
You can draw link atoms using the popup menu in two ways:
- Right-click on the atom to bring up the popup menu. Select the required
repetition number from the "Link node" submenu. Marvin will find out the
outer (non-repeating) bonds for you.
- Select the atom you would like to be the link node and two neighboring
bonds for outer (non-repeating) bonds. Right-click anywhere on the canvas
to bring up the popup menu. Select the required repetition number from the
"Link node" submenu.
Marvin will advise you if it is not possible to create a link node for the
specified configuration (for example at ring fusions).
Outer (non-repeating) bonds will be denoted by brackets crossing them, and
the repetition numbers will be put on the atom. All portion of the molecule
connected to the link atom through non-outer bonds are supposed to repeat
together with the atom. See examples below.
| Molecule with link node |
Meaning |
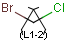 |
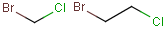 |
 |
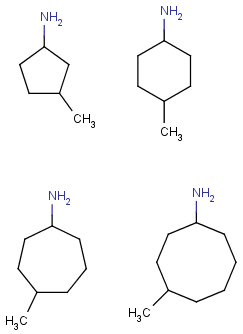 |
 |
 |
To edit a link node repetition number or change outer bonds, repeat the
drawing steps above. To make a link atom ordinary atom again, select "Off" from
the "Link node" submenu.
- Set Selection mode on by clicking
the Select button.
- To select a single atom, click
on it.
- To select two joined atoms,
click on the bond that links them.
- To select a rectangular region,
click at one corner of the desired region and drag the mouse
to the opposite corner. While the mouse button is pressed
down, a guide will be displayed to aid you.
- To select a non-rectangular
region, use the lasso select function.
- Click anywhere on the
canvas and release the mouse button.
- Draw the region with
your mouse (without pressing any mouse button). A blue
guide line will appear where you have drawn. A pink line
will connect the start and end points.
- When you have finished drawing the region, click the
mouse. In the event that you have not closed the region,
the pink line will be used to close it for you.
You can unselect all by clicking an empty area of the canvas.
Using the Erase
button:
Set Erase mode on by clicking the
Erase button.
To erase a single atom or bond,
click on it.
To erase a rectangular region,
click at one corner of the desired region and drag the mouse
to the opposite corner. While the mouse button is pressed down,
a guide will be displayed to aid you.
To select a non-rectangular
region, use the lasso erase function.
Click anywhere on the
canvas and release the mouse button.
Draw the region with
your mouse (without pressing any mouse button). A blue
guide line will appear where you have drawn. A pink line
will connect the start and end points.
When you have finished drawing the region, click
the mouse. In the event that you have not closed the
region, the pink line will be used to close it for you.
Using Selection mode:
Select a portion of the structure.
Click the Cut button or use the DELETE
button on your keyboard.
Using pop-up menus:
Right click on an atom or bond.
Select Remove from the pop-up menu.
You can edit a molecule using the methods described in
How to Draw Structures and
How to Delete a Structure.
You can alter a molecule by directly editing its source in the
Edit Source Window. You can view and edit the source in any of the
supported file formats. To change format, simply select the desired
one from the View
Menu.
To reload the molecule described by the text in this window
into the MarvinSketch canvas (including any changes you may have
made), select File >
Import As. If the automatic format
recognition detects a file format (checking it by a priority list), it will be
offered in the Select Import Mode field (Import as Recognized, indicating the file type in brackets).
If the structure is associated to a file type of higher priority than your choice,
choose the Import As option to set the file format.
For example, you want to create the seryl-asparagine dipeptide: write "SN" in the Source,
then select Import. The automatic option detects
it as SMILES, but if you select the Import As option, and then the
"Peptide Sequence" from the list, it will be imported correctly.
In addition, there are some cases when the automatic recognition cannot detect
the file format, even though the entered text is correct (although it is very
rare). In this case the Import As Recognized option is disabled and you
have to choose the format from the list of the Import As option.
Marvin allows you to clean your molecule in either 2D or 3D. Cleaning
will calculate new coordinates for the atoms. Generating conformers and
choosing the favored one is also supported.
You can initiate cleaning via the Structure
> Clean2D/3D submenu. For more information on molecule cleaning, please visit
this link.
Submenus
- Clean 2D
- Clean in 2D: cleans the molecule(s) in 2D
- Hydrogenize Chiral Cneter: adds an explicit hydrogen with a wedge bond to
chiral centers which have no terminal atoms as substituents
- Clean Wedge Bonds: changes wedge bonds for convention display
- Clean 3D
- Clean in 3D: cleans the molecule(s) in 3D
- Cleaning Method: choosing from various methods
- Display Stored Conformers: works only if conformers of the sketched molecule had
been generated with the help of the Conformer plugin, choosing the
'Store conformer information in property field' option. See details in the
plugin's documentation.
You can toggle the display of rings as aromatic using the
Structure > Aromatization submenu.
There is a wide range of functions related to the display of the
molecules. These settings can be found in the
View menu and
the Preferences dialog window.
Additionally, you can move, rotate, and zoom in/out on the structure.
You can move or rotate a selected structure.
Select the part of the structure
you wish to move.
Moving the selection:
Move the mouse pointer toward
the center of the selected structure until a blue
rectangle appears.
Translate the selection by
dragging the mouse.
Rotating the selection:
Move the mouse pointer toward
the "edge" until a blue gear appears.
Rotate the selection by dragging the mouse.
You can set the magnification of the molecule being on the canvas by the
Zoom buttons.
When a molecule is loaded into the sketcher it is scaled
automatically to fit the window.
You can set the display format for the molecule and screen
resolution using the View
> Display submenu. Available molecule formats are
Wireframe, Wireframe
with Knobs, Sticks, Ball and Stick, and Spacefill.
You can set the resolution to low or high via the Quality
submenu.
The View >Colors
submenu allows you to specify the color
scheme of the molecules. The available options are:
Marvin has a number of options for the display of implicit and
explicit hydrogens. Because Marvin is chemically intelligent, it will
automatically add hydrogens as necessary within the structure.
Generally, these will be implicit and displayed based on the options
set in the View
menu.
To view all hydrogens explicitly, displayed as atoms with bonds to
neighbors, chose Structure
> Add > Add Explicit Hydrogens. The Structure
> Remove > Remove Explicit Hydrogens will return to the previous display mode.
To view implicit hydrogens by symbol, use the View
> Misc > Implicit Hydrogens menu group. This option
is disabled in Spacefill and Ball & Stick display modes.
Displaying the label of carbon atoms in structures is possible the following way:
- Always - Always show the atom labels of carbon atoms.
- Never - Never show the atom labels of carbon atoms.
- At straight angles and at impl. Hs - Show the atom labels of carbon atoms at straight angles and at implicit Hydrogens.
This option can be set in the Display tab of the Edit
> Preferences box.
Marvin can not automatically correct all valence errors or any reaction errors. Instead,
these errors are highlighted and you may make the
appropriate corrections yourself.
This option can be enabled and disabled through the Edit
> Preferences box.
Many of the display settings in Marvin are saved and reloaded the next time you start the program.
Background color, molecule color scheme, and hydrogen visibility can be set
from the View menu and will be saved automatically when you exit the program.
Other options, including look & feel, error highlighting, and object visibility
can be set using the Preferences dialog window from
the Edit menu.
2D and 3D Viewer Windows
Choosing View >Open 2D Viewer or
Open 3D Viewer
launches a MarvinView window containing the current molecule of MarvinSketch.
More advanced display format can be obtained for the molecule by applying format styles.
Format styles in Marvin include the setting of the following attributes:
- type of atom font,
- size of atom font,
- color of atoms,
- thickness of bonds,
- color of bonds.
All these options can be collectively set using styles.
To load or define styles use the Edit > Format... menu.
This menu brings up the "Format of
the current document" dialog in which atom and bond format options can be specified.
The original attributes for
atoms and bonds can be restored by using the Reset
functions of the dialog at any time.
When loading a molecule all atoms/bonds belong to the default atom/bond set
if no styles were applied previously.
After selecting an atom/bond set and applying a style for it, the selected atoms/bonds are
removed from the default atom/bond set and a new set is created from the atoms/bonds with new style.
All the atoms/bonds, whose style were not yet modified by selection and applying a style on them,
still belong to the default atom/bond set.
Your changes might be applied for a set of atoms/bonds:
- for the selected atom/bond set,
- for the default atom/bond set,
- for all the atoms/bonds.
The top three radio buttons specify the target of the format settings being edited
in the dialog.
The "Apply changes for all the atoms/bonds" option allows loading of predefined styles
or creation of custom styles using the Load Style and Save Style buttons.
Loading a style
After pressing the Load Style button, you can load a style from a combo box or browse amongst the previously defined
style files. The chosen style will be loaded into the "Structure Drawing Properties" in the "Format of the current document" dialog.
Saving a style
Set the "Structure Drawing Properties" you wish to save and press the
Save Style button to get to the "Save" dialog where you can enter the name of the style file and save the style.
All your own saved files will be stored under the <HOMEDIR>/chemaxon/styles/ directory (where <HOMEDIR> is the user's home
directory) and will be added to the combobox items.
A new style file can be added to the chemaxon/marvin/styles directory under the Marvin installation directory.
This new style file has to be listed in file chemaxon/marvin/styles/styleFileList.properties. The new style file
will be copied to the <HOMEDIR>/chemaxon/styles/ directory and appear in the combobox of the "Loading of a journal style" dialog.
(No existing style files under <HOMEDIR>/chemaxon/styles/ are overwritten)
Return to Top
Copyright © 1998-2009
ChemAxon Ltd.
http://www.chemaxon.com/marvin