Instructions for plugin teaching
General remarks about the pKa teaching
If you are not satisfied with the performance of the default pKa calculator then
you can take the advantage of the supervised pKa learning method that is built
into the pKa calculator.
What do you need to do in order to improve the accuracy of the pKa calculation?
First, you need to see clearly which ionization center(s) was predicted
inaccurately by the pKa calculator.
You need to collect experimental data for that ionization center(s). The learning
algorithm is based on linear regression analysis, therefore you need to collect a
certain amount of experimental pKa data otherwise the regression analysis will fail.
There is no rule of thumb for what a large pool of data is required to perform a
reliable pKa teaching. If your purpose is to create a local model with the scope
only for a certain types of chemical environment of the ionization
center then it may be enough to collect a few representative structures. A more
robust model, however, requires as many as possible diverse structures and
pKa values of the ionization center in question.
The next step of the teaching process is the input of the collected data into
an sdf file. The file can be easily created by using the graphical user interface of Instant JChem.
What kind of information should be included in the sdf file?
The structure of the molecules and their experimental pKa value(s) and atomic
ID's which are assigned to the appropiate pKa value(s).
After preparing the sdf file you can run the teaching algorithm that
creates a correction library from your data. This correction library will be
used by the pKa calculation of the ionization
center in question.
Training of the pKa plugin
- Create a training set in sdfile (.sdf)
format from your experimental data. The file must
contain the following fields:
- structure
- pKa value 1 (field name: pKa1)
- ID of the atom which has the pKa1 value (field name: ID1)
Additional pKa values are optional (recommended for handling multiprotic compunds):
- pKa value 2 (pKa2)
- ID2
- etc.
Definition of only one pKa value is enough to apply the
training data, but more values in case of multiprotic compounds will enhance the
reliability of the pKa teaching.
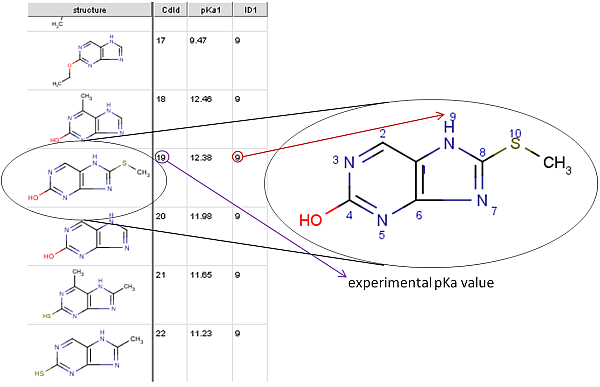
In this example this file is mydata.sdf.
The picture below is a detail from the training file. ID1 is the index
of the atom with the experimental pKa1 value (ID2 would
be the index of the second measured pKa value /pKa2/, etc.).
This atom index can be viewed by checking the Atom number option in the molecule editor (menu: View->Misc).
- Generation of the knowledge base:
Execute the following command from command line (in this example with the home directory path for Linux):
cxcalc -T pKa -o /home/myaccount/ChemAxon/MarvinBeans mydata.sdf
(option -o gives the location of the folder.)
A 'pKaReg' folder will be created containing the training data.
Create a folder called config in the Marvin installation directory.
If Marvin's default installation was followed, Marvin installation directory is located in:
- Windows: C:\Program Files\ChemAxon\MarvinBeans
- Linux: USERHOME/ChemAxon/MarvinBeans (e.g. /home/myaccount/ChemAxon/MarvinBeans)
- OS X: /Applications/ChemAxon/MarvinBeans
Copy the created pKaReg folder to the config folder.
-
Use this knowledge base via cxcalc, Chemical Terms or Marvin. The training data helps
to calculate more accurately the pKa of the
molecules and the results are closer to the experimental values.
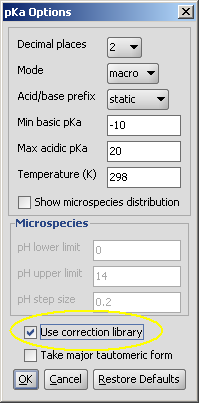
Usage in Marvin
| check the Use correction library box to activate the training option: |
 |
| pKa calculation without training data |
pKa calculation with training data |
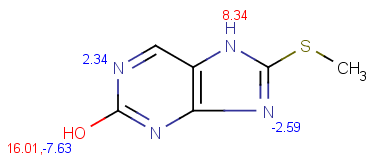
|
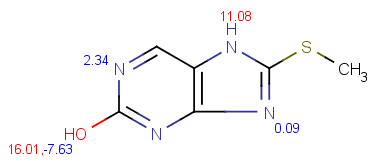
|
Usage with cxcalc
without correction library:
cxcalc pKa "CC1=NC2=C(N1)C(C)=NC(C)=N2"
id apKa1 apKa2 bpKa1 bpKa2 atoms
1 11.08 3.67 -2.38 6,9,3
with correction library:
$ cxcalc pKa -c "CC1=NC2=C(N1)C(C)=NC(C)=N2"
id apKa1 apKa2 bpKa1 bpKa2 atoms
1 9.90 3.67 -2.46 6,9,3
-c use the correction library
For more options see this page.
General remarks about the logP teaching
You can create your own logP calculator with the supervised learning method built into the logP calculator.
What you need to do is just simple collect experimental logP data and create a sdf
file from them. Details about the expected file format given below in the technical help.
What do you need to see clearly in logP model building?
If you create a local logP model then the scope of the logP calculator will be
limited. It means that the calculated logP will only provide reasonable prediction
for a few types of structures. Practically only those types of structures will be
predicted correctly which were lauched?? into the training set during the teaching process.
For example, if the training set contains only certain types of carbohydrates (carbohydrogens?) and
no other functional groups are present in the training set then it's not to expect
that the predicted logP of any amine-like structure will be accurate.
In other words, you need to be aware that a more robust general logP model requires a
large, diverse training set with thousands of structures.
Training of the logP plugin
- Create a structure file of any molecule file format from your experimental
data. The file must contain the following information:
- structure
- logP values in a property field named LOGP
In this example this file is trainingset.sdf.
- Execute the following command from command line:
cxcalc -T logP -t LOGP -o logPparameters.txt trainingset.sdf
With the -o option you can define a path for the file generated.
Create a folder called config in the Marvin installation directory
If Marvin's default installation was followed, Marvin installation directory is located in:
- Windows: C:\Program Files\ChemAxon\MarvinBeans
- Linux: USERHOME/ChemAxon/MarvinBeans (e.g. /home/myaccount/ChemAxon/MarvinBeans)
- OS X: /Applications/ChemAxon/MarvinBeans
- Save the file to the config folder with the name logPparameters.txt.
- Use this data via via cxcalc, Chemical Terms or Marvin.